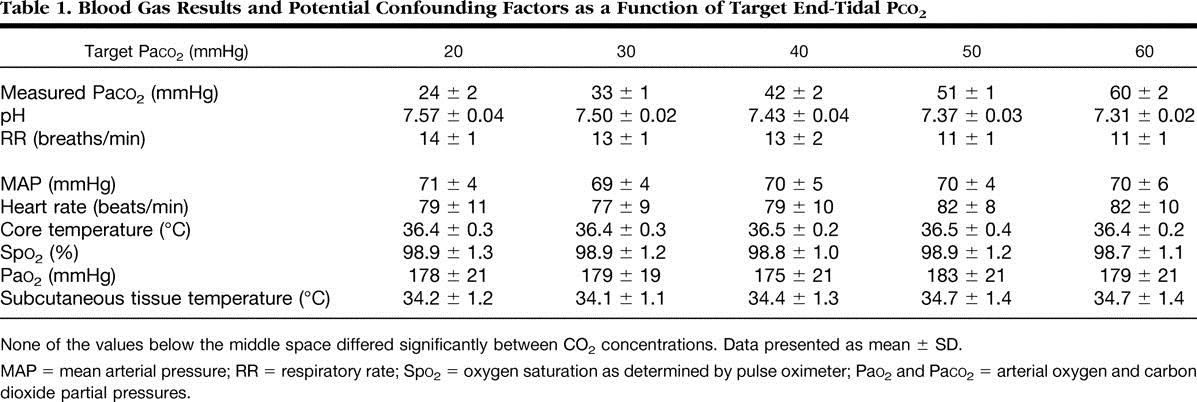
Her er en spennede studie som nevner at relasjonen mellom panikkanfall og hyperventillering kommer i utgangspunktet av metabolsk acidose, som er en kompensasjon for kronisk hyperventillering. Når CO2 synker øker nervesystemets sensitivitet til catacholaminer (stresshormoner som adrenalin og noradrenalin) og panikkanfallet kommer. I hyperkapnisk acidose (høy CO2) minker sensitiviteten til stresshormoner og kroppen beskyttes mot panikkanfall. Igjen en bekreftelse på at acidose fra CO2 er beskyttende, mens acidose fra andre ting er provoserende.
http://www.ncbi.nlm.nih.gov/pubmed/17713689
OBJECTIVE: The authors present a profile of panic disorder based on and generalized from the effects of acute and chronic hyperventilation that are characteristic of the respiratory panic disorder subtype. The review presented attempts to integrate three premises: hyperventilation is a physiological response to hypercapnia; hyperventilation can induce panic attacks; chronic hyperventilation is a protective mechanism against panic attacks.
METHOD: A selective review of the literature was made using the Medline database. Reports of the interrelationships among panic disorder, hyperventilation, acidosis, and alkalosis, as well as catecholamine release and sensitivity, were selected. The findings were structured into an integrated model.
DISCUSSION: The panic attacks experienced by individuals with panic disorder develop on the basis of metabolic acidosis, which is a compensatory response to chronic hyperventilation. The attacks are triggered by a sudden increase in (pCO2) when the latent (metabolic) acidosis manifests as hypercapnic acidosis. The acidotic condition induces catecholamine release. Sympathicotonia cannot arise during the hypercapnic phase, since low pH decreases catecholamine sensitivity. Catecholamines can provoke panic when hyperventilation causes the hypercapnia to switch to hypocapnic alkalosis (overcompensation) and catecholamine sensitivity begins to increase.
CONCLUSION: Therapeutic approaches should address long-term regulation of the respiratory pattern and elimination of metabolic acidosis.
Chronic hyperventilation predisposes an individual to PD, since compensatory mechanisms (such as alterations in renal function and tissue buffer capacity) lead to chronic metabolic acidosis, which remains latent until it is activated by chronic hypocapnia.
Therefore, therapeutic approaches should address long-term regulation of respiratory patterns60 and elimination of metabolic acidosis.