Alt om hva som gjør at vi ikke greier å holde pusten lenge.
http://ep.physoc.org/content/91/1/1.long
This article reviews the basic properties of breath-holding in humans and the possible causes of the breath at breakpoint. The simplest objective measure of breath-holding is its duration, but even this is highly variable. Breath-holding is a voluntary act, but normal subjects appear unable to breath-hold to unconsciousness. A powerful involuntary mechanism normally overrides voluntary breath-holding and causes the breath that defines the breakpoint. The occurrence of the breakpoint breath does not appear to be caused solely by a mechanism involving lung or chest shrinkage, partial pressures of blood gases or the carotid arterial chemoreceptors. This is despite the well-known properties of breath-hold duration being prolonged by large lung inflations, hyperoxia and hypocapnia and being shortened by the converse manoeuvres and by increased metabolic rate.
Breath-holding has, however, two much less well-known but important properties. First, the central respiratory rhythm appears to continue throughout breath-holding. Humans cannot therefore stop their central respiratory rhythm voluntarily. Instead, they merely suppress expression of their central respiratory rhythm and voluntarily ‘hold’ the chest at a chosen volume, possibly assisted by some tonic diaphragm activity. Second, breath-hold duration is prolonged by bilateral paralysis of the phrenic or vagus nerves. Possibly the contribution to the breakpoint from stimulation of diaphragm muscle chemoreceptors is greater than has previously been considered. At present there is no simple explanation for the breakpoint that encompasses all these properties.
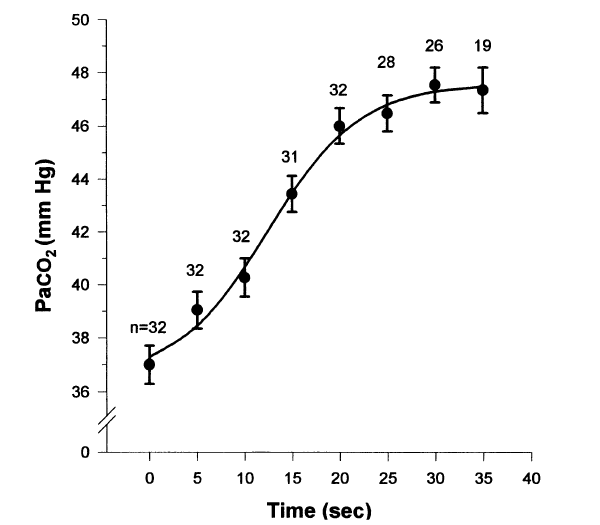
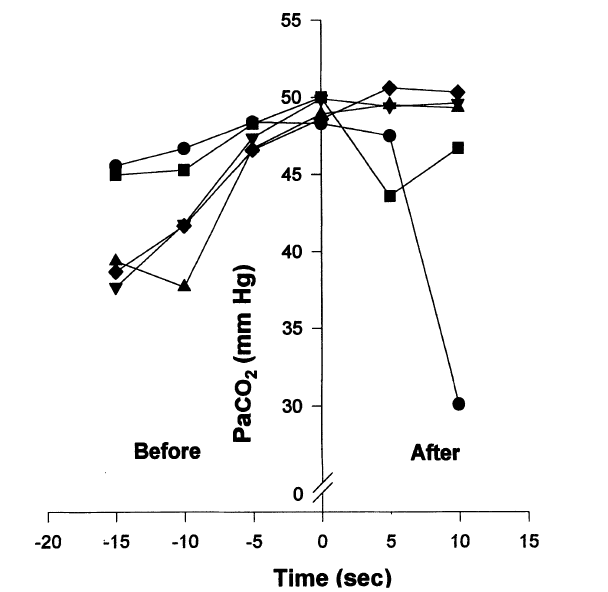
At breakpoint from maximum inflation in air, the Pa/etO2 is typically 62 ± 4 mmHg and the PetCO2 is typically 54 ± 2 mmHg (n = 5; Lin et al. 1974), and the longer the breath-hold the more they change.
Nunn (1987) estimates that consciousness in normal adults is lost at PaO2 levels below ∼27 mmHg and PaCO2 levels between 90 and 120 mmHg.
This is because the rhythmic EMG and negative pressure waves occur simultaneously, because their frequency and amplitude are within the respiratory range and because they increase as CO2 levels rise towards the end of the breath-hold. This CO2 rise would stimulate the central respiratory rhythm.