Alt om hvordan mitochondrier reagerer på hypoxi, inkludert under trening. Nevner ROS, kapillærer, oxygencascade, HIF-1,tibetanere og høydetreningmen ingen ting om CO2. Nevner også hypoxi inntrer 20s etter trening starter – ref HIIT.
http://ep.physoc.org/content/88/1/109.long
In contrast to earlier assumptions it is now established that permanent or long-term exposure to severe environmental hypoxia decreases the mitochondrial content of muscle fibres. Oxidative muscle metabolism is shifted towards a higher reliance on carbohydrates as a fuel, and intramyocellular lipid substrate stores are reduced. Moreover, in muscle cells of mountaineers returning from the Himalayas, we find accumulations of lipofuscin, believed to be a mitochondrial degradation product. Low mitochondrial contents are also observed in high-altitude natives such as Sherpas.
In these subjects high-altitude performance seems to be improved by better coupling between ATP demand and supply pathways as well as better metabolite homeostasis.
The hypoxia-inducible factor 1 (HIF-1) has been identified as a master regulator for the expression of genes involved in the hypoxia response, such as genes coding for glucose transporters, glycolytic enzymes and vascular endothelial growth factor (VEGF). HIF-1 achieves this by binding to hypoxia response elements in the promoter regions of these genes, whereby the increase of HIF-1 in hypoxia is the consequence of a reduced degradation of its dominant subunit HIF-1a.
A further mechanism that seems implicated in the hypoxia response of muscle mitochondria is related to the formation of reactive oxygen species (ROS) in mitochondria during oxidative phosphorylation. How exactly ROS interfere with HIF-1a as well as MAP kinase and other signalling pathways is debated.
The current evidence suggests that mitochondria themselves could be important players in oxygen sensing.
The focus on mitochondria neglects the fact that for the dominant function of mitochondria, aerobic ATP re-synthesis, the mitochondrial organelle cannot function independently. It is embedded, at least in animal phyla, in a systems physiological context. Mitochondria house the final biochemical steps in the production of reducing equivalents that react at the terminal oxidases of the respiratory chain with molecular oxygen provided by the respiratory system from the environment.
In mitochondria, O2 finally disappears and oxygen partial pressure goes to zero (Fig. 1). Mitochondria thus are an effective oxygen sink and this allows organisms to use all of the available oxygen partial pressure of the actual environment to drive the respiratory cascade from lungs through circulation to the mitochondria (Taylor & Weibel, 1981).
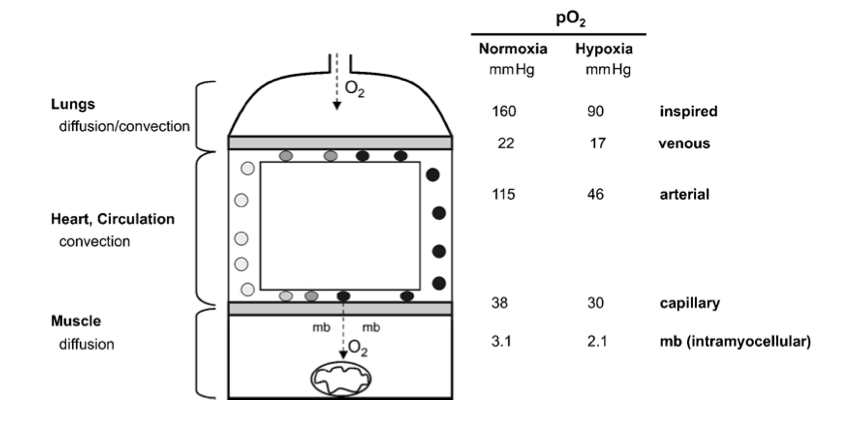
Figure 1
The pathway of oxygen. Simplified model of the oxygen transport pathway showing the principal structures and the corresponding partial pressure of oxygen (PJ) in these compartments, during maximal exercise, while breathing normoxic or hypoxic room air. mb, myoglobin. The data are derived from Richardson et al. (1995).
What is known is that isolated mitochondria do not need more than a few torr of O2 partial pressure for full function (Gayeski & Honig, 1997). Similarly, it is generally acknowledged that assessing mitochondrial function in hypoxia in cultured cells requires the lowering of the oxygen pressure at the cell surface to values between 0.5 and 5 Torr, values completely incompatible with the survival of all but a few highly specialized species (Boutilier & St Pierre, 2002).
In the context of this review we mostly neglect the hypoxic response of the microvasculature as well as the cardio- vascular and pulmonary system, which all conspire to maintain mitochondrial function under hypoxic conditions (Hochachka, 1998).
With ‘short- term’ hypoxia we refer to continuous bouts of hypoxia exposure lasting from minutes to hours (such as during a physical exercise bout), whereby during most of the day subjects are subjected to normoxic conditions.
With the term ‘intermittent’ hypoxia we designate experimental protocols whereby short periods of hypoxia (minutes) are interspersed with similarly short periods of normoxia. The physiological effects of inter- mittent hypoxia interventions have not yet been well defined. In particular, these types of interventions have not to our knowledge been analysed with regard to changes of the mitochondrial compartment. This literature will thus not be reviewed here (see Serebrovskaya, 2002).
One of the first notions of a change of muscle oxidative capacity with hypoxia is found in Reynafarje’s (1962) landmark paper on muscle adaptations in Peruvian miners. He found cytochrome c reductase activity (+78 %) and myoglobin content (+16%) to be significantly increased in biopsies of sartorius muscle of permanent high-altitude residents as compared to low-landers. This paper, together with data of larger muscle capillary densities in guinea pigs native to the Andeas as compared to data from low-land control animals (Valdivia, 1958), had a strong impact on physiologists’ thinking for at least two decades.
When it became clear towards the end of the sixties, that endurance-type exercise was also a potent stimulator of muscle mitochondrial enzyme activity (Holloszy, 1967), the connection was made that local tissue hypoxia provoked by exercise might be an important signal for mitochondrial proliferation under heavy exercise conditions.
The capillary density (i.e. the number of capillaries counted per unit area of muscle fibre) was indeed found to be increased in post-expedition samples. However, this was not due to capillary neo-formation since the capillary to muscle fibre ratio was unchanged. What had occurred over the time of the expedition was a significant loss of muscle fibre cross- sectional area. Hence, the same number of capillaries supplied a smaller tissue space. In addition, we found a decrease of mitochondrial volume density, which led to a total loss of muscle mitochondrial volume of close to 30 %.
This adaptation was suggested to represent an anti-apoptotic mechanism allowing protection against the lack of oxygen in oxidative muscles (Riva et al. 2001).
We would assume that the increased transcription of genes involved in the metabolic pathways favouring glucose metabolism in muscle cells is responsible for the observed shift towards glucose metabolism in organisms at altitude. The functional advantage of this adaptation would be a larger ATP yield per molecule of oxygen (Wilmore & Costill, 1994). It has recently been shown that protein expression of the enzymes and transporters of lactate, lactate dehydrogenase (LDH) and monocarboxylate transporters (MCT1, 2 and 4), is affected in a tissue-specific manner by long-term exposure to hypobaric hypoxia (McClelland & Brooks, 2002). These findings are broadly compatible with the idea that hypoxia induces mechanisms favouring glucose utilization in muscle cells.
Despite the low muscle mitochondrial oxidative capacity, physical performance capacity of native high-land populations at altitude is excellent. A number of physiological mechanisms have been invoked to explain the ‘paradoxical’ finding of superior aerobic performance in hypoxia despite modest muscle oxidative capacities (Hochachka et al. 2002). Hochachka et al. (1998) describe systemic mechanisms, among them being a blunted hypoxia ventilatory and pulmonary vasoconstrictor response as well as an up-regulation of erythropoietin expression, which help native high-land populations to excel at altitude. At the level of muscle tissue it is suggested that the control contributions from cellular ATP demand and ATP supply pathways are up-regulated whereas the contribution of control steps in O2 delivery are down-regulated (Hochachka et al. 2002). This leads to an improved coupling between ATP demand and supply pathways and better metabolite (i.e. lactate, adenylates) homeostasis.
We thus decided to explore hypoxia protocols under which hypoxia was present only during the exercise session but not during recovery (e.g living low–training high). Analysis of pre- and post- training biopsies of m. vastus lateralis revealed that total mitochondria increased significantly in all groups; in contrast, subsarcolemmal mitochondria, i.e. those located near capillaries, increased significantly only in those groups training under hypoxic conditions, irrespective of training intensity. Noticeably, the group which trained at high intensity in hypoxia showed the highest increase in total mitochondrial volume density (+59 %) and capillary length density was increased significantly in this group only (+17.2 %) (Geiser et al. 2001; Vogt et al. 2001).
Several independent lines of evidence suggest the occurrence of ‘local hypoxia’, i.e. a drop in intramyocellular oxygen pressure with exercise.
This finding of a low muscle oxygen tension during exercise is supported by more recent data using 1H-nuclear magnetic resonance spectroscopy which demonstrates that myoglobin desaturation occurs within 20 s of onset of exercise in human quadriceps muscle (Richardson et al. 1995).
Mitochondria, the oxygen sink, are more prevalent in oxidative fibres by a factor of at least three, and even within fibres they are clustered in the fibre periphery close to the capillaries (Howald et al. 1985).
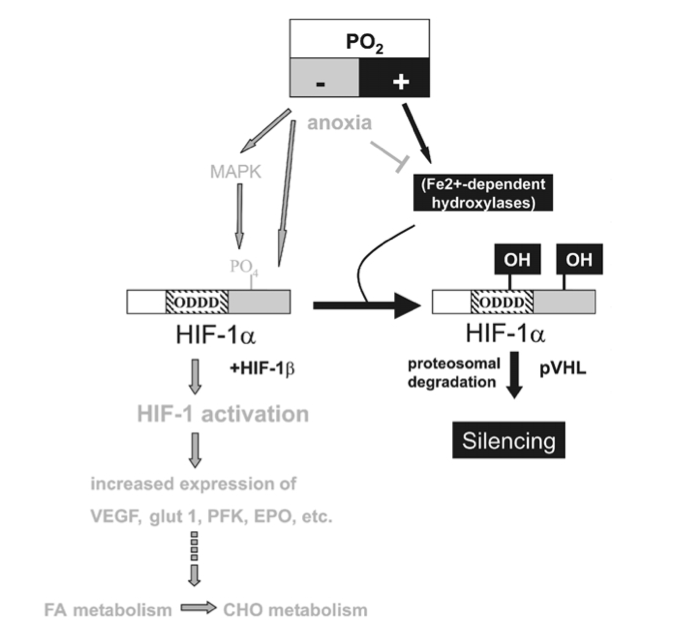
Figure 3
HIF-1a-mediated oxygen sensor and hypoxia-inducible gene expression. Oxygen stabilizes hypoxia- inducible factor 1a (HIF-1a) through Fe2+-dependent proline hydroxylases and asparaginyl hydroxylase(s) which hydroxylate HIF-1a within its oxygen-dependent degradation domain (ODDD) and C-terminal portion, respectively. Such modification causes recruitment of the von Hippel-Lindau tumour- suppressor protein (pVHL), which targets HIF-1a for proteosomal degradation thereby silencing HIF-1a activity. Conversely, hypoxia causes a stabilization of HIF-1a and activates the MAPK pathway which enhances the transcriptional activity of HIF-1a through phosphorylation. Both mechanisms contribute to transcriptional activation of downstream angiogenic factor (VEGF), erythropoietin (EPO), glucose transporters (glut 1 and 3) and glycolytic genes (PFK) via HIF-1a/HIF-1b dimers (HIF-1). Moreover, lack of oxygen (anoxia) causes HIF-1a stabilization, possibly by reducing the activity of hydoxylases by depleting their substrate O2.
In vitro experiments show that an activation of HIF-1 leads to an up-regulation of anaerobic enzymes of the glycolytic pathway (Wenger, 2002).
To date there is no clear-cut explanation for the paradoxical finding of increased reactive oxygen species (ROS) production in hypoxia as there is controversy about whether hypoxia, in fact, causes an increase or a fall in ROS production (Archer & Michelakis, 2002). Also the mechanisms by which ROS and cytosolic H2O2 levels are involved in stabilization/activation and eventual degradation/silencing of HIF-1a and other redox-sensitive transcription factors are controversially discussed (Chandel et al. 2000; Kietzmann et al. 2000).
It is reported that ~2% of the O2 used by the mitochondrial electron transport chain creates ROS and in particular the superoxide radical, due to its incomplete reduction (see Kietzmann et al. 2000; Archer & Michelakis, 2002; reviewed in Abele et al. 2002). The superoxide anion (O2•_) radical is very unstable and is rapidly converted either spontaneously or after its export into the cytoplasm by mitochondrial and cytoplasmic superoxide dismutases, MnSOD and Cu/ZnSOD, to the more stable H2O2.
Recent findings in hepatoma cells indicate a small (approximately 2.5-fold) and transient increase in ROS after 40–50 min of hypoxia onset (0.5 % O2; Vanden Hoek et al. 1998). These and subsequent in vitro experiments in the same system point to mitochondria, and to respiratory complex III of the electron transport chain in particular, as the source of ROS production in hypoxia (Chandel et al. 2000).
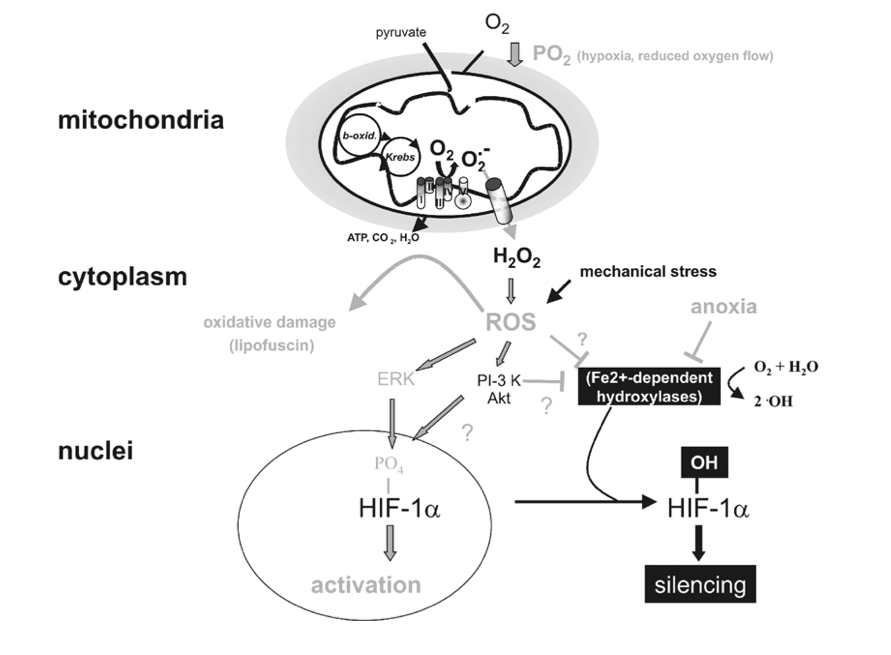
Figure 4
Targets of mitochondrial ROS production. Incomplete reduction of O2 during normal metabolic conversion of pyruvate in mitochondria gives rise to a low level of superoxide anion (O2•_). Exposure of cells to hypoxia increases the aberrant production of O2•_ at the mitochondrial electron chain complex III. Catalysed or spontaneous dismutation of the unstable superanion and export via anion channels enhances the concentration of cytosolic H2O2 and other reactive oxygen species (ROS). Increased ROS may increase the level of oxidatively damaged lipids (lipofuscin) or activate downstream redox-sensitive signal transduction events. For example ROS stabilize HIF-1a possibly by interfering with the activity of Fe2+-dependent proline hydroxylases. ROS may also activate the MAPK or PI-3K/Akt pathway, which enhance the transcriptional activity of HIF-1a through its phosphorylation. Lastly, mechanical stress may also increase ROS production which may influence HIF-1a activation.
Clearly there is evidence pointing to a role of mitochondria as oxygen sensors by modulation of ROS production.
Recently, evidence for increased oxidant production in rat skeletal muscle during prolonged exercise has been provided, with both the mitochondrial respiratory chain and the NADPH oxidase as potential sources for oxidants (Bejma & Ji, 1999). This argues for ROS production in skeletal muscle due to local tissue hypoxia.
