


Viktig studie som hinter til den virkelige oppgaven til nervesystemet, som ikke er sende info inn til eller ut fra hjernen… Om vitenskapens forstelse av nervesystemet opp igjennom tidene og hvordan alt vi trodde om nervesystemet er feil.
http://adb.sagepub.com/content/21/2/67.abstract
Hele studien på dropbox: https://dl.dropboxusercontent.com/u/17457302/Forskning%20mappe%20for%20terapi/Keizer%202013%20What%20nervous%20systems%20do-%20early%20evolution%2C%20input%C2%96output%2C%20and%20the%20skin%20brain%20thesis.pdf
We hold that the fundamental problem here was not so much to act intelligentlya problem that had already been solved in various ways without a nervous system (Section 3.3)but to act as a single multicellular unit.
Nervous systems arose as a source and coordinator of patterned activity across extensive areas of contractile tissue in a way that was only loosely constrained by sensor activity.
In this view, the central direction of nervous system connections runs transverseat right anglesto the through-conducting stream that runs between sensors and effectors: early nervous systems evolved as connec- tions across a contractile tissue and in close connection to the animal epithelium or skin.
Adopting the phrase skin brain introduced by Holland (2003), we will refer to this idea as the skin brain thesis, or SBT.
Although the inputoutput view is deeply entrenched, there are issues involving nervous system functioning that are highly puzzling or awkward when the input output view is taken as a fundamental account of ner- vous systems.
The current inputoutput interpretation of nervous sys- tems is closely linked to a computational information- processing interpretation. This linkage is intrinsic to the classic neuron doctrine, according to which neurons are individual entities that receive and send electrical signals to one another through synapses in an all-or-none fashion that is basically similar to electrical switches. Consistent with the neuron doctrines one-way flow of information, nervous systems could be interpreted as electronic circui- try, which may be far more complex than artificial circui- try, but not intrinsically different.
The problem with this input-output interpretation is that the neuron doctrine on which it is based has been seriously undermined (e.g., Bullock et al., 2005; Guillery, 2007; Kruger & Otis, 2007) since it was first advanced by Ramon y Cajal in the late 19th century. Famously, Cajal formulated what came to be called the neuron doctrine explicitly in opposition to the then-current idea that ner- vous systems are reticular organizations of nerve cells directly connected to one another, through which electri- cal activity flows diffusely in all directions (Guillery, 2007; Kruger & Otis, 2007).
The neu- ron doctrine can not plausibly explain the diversity of neuromodulatory substances, such as amines and neu- ropeptides, that remodel neuron behavior and circuitry within minutes and hours instead of the standard milli- second time scale (Bullock et al., 2005). Many of these neuromodulatory molecules are not recent evolutionary developments but have a deep genomic history. More recently, immune system elements, such as cytokines, have been shown to play critical roles in modulating neural plasticity under normal as well as challenged conditions (McAfoose and Baune, 2009; Yirmiya and Goshen, 2011), and these associations are also very old (Maier and Watkins, 1998). The neuron doctrine cannot explain these associations either. Moreover, in many neurons, action potentials can travel backward from the axon and cell body to the dendrites.
Clue 2: The detailed operation of neurons and nervous sys- tems is much more complex and diverse than can be readily accounted for by the inputoutput view.
Clue 3: The reflex arc organization may very well be a sec- ondary optimization of nervous systems.
The inputoutput interpretation stresses that nervous systems function as information processing devices. However, in recent years serious claims concerning the complexity, and even cognitive, nature of the behavior of single-celled organisms have come to the fore. For example, John Allman (1999) discusses how the most fundamental features of brains such as sensory integra- tion, memory, decision-making, and the control of behavior, can already be found in simple organisms such as bacteria (pp. 56).
While this is presumably true of complex ner- vous systems, the point does not seem to apply to basic forms. When one systematically compares organisms with basic nervous systems, they do not show more complex behavior than creatures without a nervous sys- tem.
According to Jennings, the possession of a nervous system brings with it no observable essential changes in the nature of behavior. We have found no important additional features in the behavior when the nervous system is added (p. 263).
Clue 4: Basic nervous systems do not lead to more complex behavior than is often present in organisms without a nervous system.
Clue 5: Many of the biomolecular characteristics of neurons are already present in non-neural precursor contexts.
Clue 6: Understanding what nervous systems do is a question that requires an answer at the level of the whole animal.
Clue 7: The main animal effector consists of muscle tissue that requires spatiotemporal coordination.
Clue 8: Coordinating extensive areas of muscle tissue requires endogenous activity.
Nowadays, the picture has changed again. While Mackies scenario for the origins of nervous systems is still influential (e.g., Arendt, 2008; Je kely, 2011; Miller, 2009), it faces important difficulties. A key problem is that nervous systems are found more widely among animal phyla and classes than electri- cally coupled conductive epithelia. Notably, while all four major cnidarian classes have a nervous system, there is substantial evidence that only the Hydrozoa have functional gap junctions (Mackie, Anderson, & Singla, 1984; Satterlie, 2011).
Clue 9: Chemical transmission between adjacent cells can have provided the basis for primitive conductive epithelia that formed a half-way station to nerve nets.
Clue 10: Chemically transmitting conductive (myo)epithelia can have provided a basic form of muscle coordination.
Clue 11: Specialized axodendritic connections can have sub- sequently evolved to broaden the existing possibilities for muscle coordination.
Under this interpretation, the core business of such nerve nets consisted of organizing and integrating activity across contractile effector surfaces (e.g., mus- cle) spread out beneath an external epithelium. Such a task would involve parallel organization and coordina- tion requiring signaling across a surface rather than a through-conducting, sequential organization based on a set of pre-existing sensors and effectors. No stimulus can specify by itself the behaviorally relevant contrac- tion patterns across such a surface. Patterns that workthat is, patterns that lead to movements that are appropriate under the circumstancesare a func- tion of the particular effector surface that is present in the animals rather than of any triggering stimulus. Also, based on what we know about organisms today, movement is likely to have been self-induced, while external stimuli acted rather as modulating factors on continuous effector activity.
While modern nervous sys- tems have various other functions, it is evident that enabling an organism to move and manipulate its envi- ronment in specific ways is the prime reason for the huge investment in these metabolically expensive organs (Allman, 1999).
Such cellular con- tractions must be coordinated with respect to one another, however. Uncoordinated contractions by indi- vidual cells would not result in whole-body motility. This, we believe, is where nervous systems come in. Nerve nets are intrinsically tied up with muscle surfaces.
The SBT can now be formulated as the proposition that early nerve nets evolved when some conducting cellseither within or connected to the myoepithelium evolved elongated processes and synaptic connections in a way that modified and enhanced the patterning capabil- ities of a pre-existing myoepithelium. Rather than pro- viding specific connections from sensors to effectors, the proper function of such nerve nets was to control, modify and extend the available self-organized pattern- ing across a Pantin surface. The key adaptation pro- vided by early nerve nets was the way in which they added to the generic self-organizing properties of pre- existing epithelial and muscular tissues.
To summarize, the SBT claims that nerve nets origi- nated as a new mechanism by which Pantin surfaces could be more intricately and flexibly patterned to accommodate efficient motility at larger bodily scales. At a fundamental level nerve nets are fitted to spatial patterning and to accommodating spatially patterned feedback.
The SBT offers a genuinely new conceptual approach for understanding nervous systems at a whole systems level. Starting with the most primitive neural organiza- tionsproto-neural myoepithelia and nerve netswe argue that both are characterized by connections trans- verse to the standard sensor-effector direction and evolved their characteristics to bind the many cellular units of muscle sheets together into a unitary system. Nervous systems are in this view not organized aroundor rather betweensensors and effectors. They are themselves a precondition for both extended con- tractile effectors as well as multicellular sensory arrays.
We have stressed from the beginning that the SBT provides a conceptual reinterpretation of nervous system functioning.
The skin brain pro- posal casts animal behavior as a dynamical phenotype, necessarily tied to the species or class of animals under consideration. Sherrington once observed that posture follows movement like a shadow (Stuart, 2005). We would like to stress that dynamically changing body pos- ture is a precondition for all task-oriented animal beha- vior. Animal behavior is a part of animal organization.
Om CO2 i helbredelse av vev. Viktig oversikt som nevner CO2 sin bane og effekt gjennom hele organismen – fra DNA til celle til vev til blod. Bekrefter ALT jeg har funnet om CO2 og pusteknikkene. Nevner også potensiell farer, som kun skjer ved akutt hypercapnia. Nevner også en meget spennende konsept om å buffre CO2 acidose med bikarbonat (Natron). I kliniske tilfeller på sykehus kan det ha negative effekter, men hos normale mennesker vil det virke som en effektiv buffer.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2887152/
Hypercapnia may play a beneficial role in the pathogenesis of inflammation and tissue injury, but may hinder the host response to sepsis and reduce repair. In contrast, hypocapnia may be a pathogenic entity in the setting of critical illness.
For practical purposes, PaCO2 reflects the rate of CO2 elimination.
The commonest reason for hypercapnia in ventilated patients is a reduced tidal volume (VT); this situation is termed permissive hypercapnia.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2887152/table/T1/?report=previmg
High VT causes, or potentiates, lung injury [4]. Smaller VT often leads to elevated PaCO2, termed permissive hypercapnia, and is associated with better survival [5,6]. These low-VT strategies are not confined to patients with acute lung injury (ALI)/acute respiratory distress syndrome (ARDS); they were first reported successful in severe asthma [7], and attest to the overall safety of hypercapnia. Indeed, hypercapnia in the presence of higher VT may independently improve survival [8].
Hypocapnia is common in several diseases (Table (Table1;1; for example, early asthma, high-altitude pulmonary edema, lung injury), is a common acid-base disturbance and a criterion for systemic inflammatory response syndrome [9], and is a prognostic marker of adverse outcome in diabetic ketoacidosis [10]. Hypocapnia – often prolonged – remains common in the management of adult [11] and pediatric [12] acute brain injury.
Causes of hypocapnia
| Hypoxemia | Altitude, pulmonary disease |
| Pulmonary disorders | Pneumonia, interstitial pneumonitis, fibrosis, edema, pulmonary emboli, vascular disease, bronchial asthma, pneumothorax |
| Cardiovascular system disorders | Congestive heart failure, hypotension |
| Metabolic disorders | Acidosis (diabetic, renal, lactic), hepatic failure |
| Central nervous system disorders | Psychogenic/anxiety hyperventilation, central nervous system infection, central nervous system tumors |
| Drug induced | Salicylates, methylxanthines, β-adrenergic agonists, progesterone |
| Miscellaneous | Fever, sepsis, pain, pregnancy |
CO2 is carried in the blood as HCO3-, in combination with hemoglobin and plasma proteins, and in solution. Inside the cell, CO2 interacts with H2O to produce carbonic acid (H2CO3), which is in equilibrium with H+ and HCO3-, a reaction catalyzed by carbonic anhydrase. CO2 transport into cells is complex, and passive diffusion, specific transporters and rhesus proteins may all be involved.
CO2 is sensed in central and peripheral neurons. Changes in CO2 and H+ are sensed in chemosensitive neurons in the carotid body and in the hindbrain [13,14]. Whether CO2 or the pH are preferentially sensed is unclear, but the ventilatory response to hypercapnic acidosis (HCA) exceeds that of an equivalent degree of metabolic acidosis [15], suggesting specific CO2 sensing.
An in vitro study has demonstrated that elevated CO2 levels suppress expression of TNF and other cytokines by pulmonary artery endothelial cells via suppression of NF-κB activation [18].
Furthermore, hypercapnia inhibits pulmonary epithelial wound repair also via an NF-κB mechanism [19].
The physiologic effects of CO2 are diverse and incompletely understood, with direct effects often counterbalanced by indirect effects.
Hypocapnia can worsen ventilation-perfusion matching and gas exchange in the lung via a number of mechanisms, including bronchoconstriction [21], reduction in collateral ventilation [22], reduction in parenchymal compliance [23], and attenuation of hypoxic pulmonary vasoconstriction and increased intrapulmonary shunting [24].
CO2 stimulates ventilation (see above). Peripheral chemoreceptors respond more rapidly than the central neurons, but central chemosensors make a larger contribution to stimulating ventilation. CO2increases cerebral blood flow (CBF) by 1 to 2 ml/100 g/minute per 1 mmHg in PaCO2[25], an effect mediated by pH rather than by the partial pressure of CO2.
Hypercapnia elevates both the partial pressure of O2 in the blood and CBF, and reducing PaCO2 to 20 to 25 mmHg decreases CBF by 40 to 50% [26]. The effect of CO2 on CBF is far larger than its effect on the cerebral blood volume. During sustained hypocapnia, CBF recovers to within 10% baseline by 4 hours; and because lowered HCO3-returns the pH towards normal, abrupt normalization of CO2 results in (net) alkalemia and risks rebound hyperemia.
Hypocapnia increases both neuronal excitability and excitatory (glutamatergic) synaptic transmission, and suppresses GABA-A-mediated inhibition, resulting in increased O2 consumption and uncoupling of metabolism to CBF [27].
Hypercapnia directly inhibits cardiac and vascular muscle contractility, effects that are counterbalanced by sympathoadrenal increases in heart rate and contractility, increasing the cardiac output overall [28].
Indeed, a large body of evidence now attests to the ability of hypercapnia to increase peripheral tissue oxygenation, independently of its effects on cardiac output [30,31].
The beneficial effects of HCA in such models are increasingly well understood, and include attenuation of lung neutrophil recruitment, pulmonary and systemic cytokine concentrations, cell apoptosis, and O2-derived and nitrogen-derived free radical injury.
Concern has been raised regarding the potential for the anti-inflammatory effects of HCA to impair the host response to infection. In early pulmonary infection, this potential impairment does not appear to occur, with HCA reducing the severity of acute-severe Escherichia coli pneumonia-induced ALI [41]. In the setting of more established E. coli pneumonia, HCA is also protective [42].
Hypocapnia increases microvascular permeability and impairs alveolar fluid reabsorption in the isolated rat lung, due to an associated decrease in Na/K-ATPase activity [47].
HCA protects the heart following ischemia-reperfusion injury.
Hypercapnia attenuates hypoxic-ischemic brain injury in the immature rat [52] and protects the porcine brain from reoxygenation injury by attenuation of free radical action. Hypercapnia increases the size of the region at risk of infarction in experimental acute focal ischemia; in hypoxic-ischemic injury in the immature rat brain, hypocapnia worsens the histologic magnitude of stroke [52] and is associated with a decrease in CBF to the hypoxia-injured brain as well as disturbance of glucose utilization and phosphate reserves.
Indeed, hypocapnia may be directly neurotoxic, through increased incorporation of choline into membrane phospholipids [56].
Rapid induction of hypercapnia in the critically ill patient may have adverse effects. Acute hypercapnia impairs myocardial function.
In patients managed with protective ventilation strategies, buffering of the acidosis induced by hypercapnia remains a common – albeit controversial – clinical practice.
While bicarbonate may correct the arterial pH, it may worsen an intracellular acidosis because the CO2 produced when bicarbonate reacts with metabolic acids diffuses readily across cell membranes, whereas bicarbonate cannot.
Hypocapnia is an underappreciated phenomenon in the critically ill patient, and is potentially deleterious, particularly when severe or prolonged. Hypocapnia should be avoided except in specific clinical situations; when induced, hypercapnic acidosis should be for specific indications while definitive measures are undertaken.
Nevner det meste om gluten relatert til nerveproblemer. Noe av årsaken ligger i at det blir en autoimmun plage med antistoffer i nerver, som etter hvert gir nerveskader. 64% av de med cøliaki har også nevropati.
Hypothesis: Gluten causes symptoms, in both celiac disease and non-celiac gluten-sensitivity, by its adverse actions on the nervous system.
Many celiac patients experience neurological symptoms, frequently associated with malfunction of the autonomic nervous system. These neurological symptoms can present in celiac patients who are well nourished. The crucial point, however, is that gluten-sensitivity can also be associated with neurological symptoms in patients who do not have any mucosal gut damage (that is, without celiac disease).
Gluten can cause neurological harm through a combination of cross reacting antibodies, immune com- plex disease and direct toxicity. These nervous system affects include: dysregulation of the autonomic nervous system, cerebella ataxia, hypotonia, developmental delay, learning disorders, depression, migraine, and headache.
If gluten is the putative harmful agent, then there is no requirement to invoke gut damage and nutri- tional deficiency to explain the myriad of the symptoms experienced by sufferers of celiac disease and gluten-sensitivity. This is called ‘‘The Gluten Syndrome”.
A mechanism for such nerve damage might be through autoimmune damage [15]. A number of nerve and brain antibodies have been detected. Anti-ganglioside antibodies have been detected in 64% of patients with celiac disease who had also been troubled with some sort of neuropathy [16]. These auto-antibodies have been shown to bind to a number of critical nerve sites that will go on to damage the nerve.
Svært viktig studie med alt om smerte, fra Melzaks Body-Self Neuromatrix. Smerteforståelsens historie, fantomsmerter, hypersensitivitet, nervedegenerasjon, betennelser, Gate Control og Neuromatrix teori, m.m. Her forklares hvordan kroppsopplevelsen skapes i hjernen, selv uten noen input fra kroppen. Nevner også at smerte kan sette seg som et minne; somatic memory. Og vier mye plass til hvordan stress og kortisol bidrar til kroniske smerter, muskelsvikt og nedsatt immunsystem.
http://onlinelibrary.wiley.com/doi/10.1002/wcs.1201/full
Smerte har en funksjon i menneskekroppen som har utviklet seg i evolusjonen for å holde oss i live. Det gjør at vi tilpasser vår aktivitet så kroppen kan fokusere på helbredelse.
Pain has many valuable functions. It often signals injury or disease, generates a wide range of adaptive behaviors, and promotes healing through rest.
Men de siste 100-årenes (og foreløpige) forståelse av selve årsaken til smerte, hvordan den oppstår og hvordan den forsvinner, er basert på et mekanisk kroppsbilde som ikke tar hensyn til den subjektive smerteopplevelse. Melsaks arbeid viser oss hvordan vi snur dette og får en bedre og mer korrekt forståelse av smertefunksjonen:
Pain is a personal, subjective experience influenced by cultural learning, the meaning of the situation, attention, and other psychological variables. Pain processes do not begin with the stimulation of receptors. Rather, injury or disease produces neural signals that enter an active nervous system that (in the adult organism) is the substrate of past experience, culture, and a host of other environmental and personal factors.
Pain is not simply the end product of a linear sensory transmission system; it is a dynamic process that involves continuous interactions among complex ascending and descending systems. The neuromatrix theory guides us away from the Cartesian concept of pain as a sensation produced by injury, inflammation, or other tissue pathology and toward the concept of pain as a multidimensional experience produced by multiple influences.
Smerte er en helbredelsesfunksjon. Den hjelper oss å unngå truende situasjoner og sørger for at vi gir kroppen mulighet til å helbrede seg. Det er en naturlig og intelligent biologisk funksjon som i milliarder av år igjennom evolusjonen har sørget for at vi overlever så lenge som mulig.
We all know that pain has many valuable functions. It often signals injury or disease and generates a wide range of behaviors to end it and to treat its causes. Chest pain, for example, may be a symptom of heart disease, and may compel us to seek a physician’s help. Memories of past pain and suffering also serve as signals for us to avoid potentially dangerous situations. Yet another beneficial effect of pain, notably after serious injury or disease, is to make us rest, thereby promoting the body’s healing processes. All of these actions induced by pain—to escape, avoid, or rest—have obvious value for survival.
Smerteproblematikk har eksplodert de siste 20-30 årene og korsryggsmerter har overtatt plassen fra sult som den viktigste årsaken til ubehag blandt verdens befolkning. Melzak foreslår at vi bør se på kronisk smerte som en sykdom i seg selv, ikke som et symptom. En sykdom som følge av at nervesystemets alarm-mekanismer har slått seg vrang.
The pain, not the physical impairment, prevents them from leading a normal life. Likewise, most backaches, headaches, muscle pains, nerve pains, pelvic pains, and facial pains serve no discernible purpose, are resistant to treatment, and are a catastrophe for the people who are afflicted.
Pain may be the warning signal that saves the lives of some people, but it destroys the lives of countless others. Chronic pains, clearly, are not a warning to prevent physical injury or disease. They are the disease—the result of neural mechanisms gone awry.1–3
A BRIEF HISTORY OF PAIN
I smerteforskning og forståelse har vi, siden Descartes tid på 1600-tallet, beveget oss fra utsiden av kroppen igjennom det vi trodde var smerte-nervetråder, inn til ryggmargens «Gate Control», og nå, med The Neuromatrix, kommet opp til selve hjernen hvor vår opplevde virkelighet faktisk skapes. Først nå de siste årene har vi begynt å inkludere hjernens forskjellige funksjoner og dens eget «bilde» og opplevelse av kroppen. Tidligere ville pasienter som ikke ble bedre av kirurgi eller behandling bare bli avfeid av legene og heller sent til psykolog, hvor de heller ikke fikk noe spesifikk hjelp for smertene. Først nå, endelig, kan behandling av kronisk smerte inkludere større deler av mennesket som stemmer bedre overens med realiteten i både den subjektive opplevelsen og den vitenskapelige forklaringsmodellen.
The theory of pain we inherited in the 20th century was proposed by Descartes three centuries earlier. The impact of Descartes’ specificity theory was enormous. It influenced experiments on the anatomy and physiology of pain up to the first half of the 20th century (reviewed in Ref 4). This body of research is marked by a search for specific pain fibers and pathways and a pain center in the brain. The result was a concept of pain as a specific, direct-line sensory projection system. This rigid anatomy of pain in the 1950s led to attempts to treat severe chronic pain by a variety of neurosurgical lesions. Descartes’ specificity theory, then, determined the ‘facts’ as they were known up to the middle of the 20th century, and even determined therapy.
Specificity theory proposed that injury activates specific pain receptors and fibers which, in turn, project pain impulses through a spinal pain pathway to a pain center in the brain. The psychological experience of pain, therefore, was virtually equated with peripheral injury. In the 1950s, there was no room for psychological contributions to pain, such as attention, past experience, anxiety, depression, and the meaning of the situation.
Patients who suffered back pain without presenting signs of organic disease were often labeled as psychologically disturbed and sent to psychiatrists.
However, in none of these theories was there an explicit role for the brain other than as a passive receiver of messages. Nevertheless, the successive theoretical concepts moved the field in the right direction: into the spinal cord and away from the periphery as the exclusive answer to pain. At least the field of pain was making its way up toward the brain.

(D) Gate control theory. The large (L) and small (S) fibers project to the substantia gelatinosa (SG) and first central transmission (T) cells. The central control trigger is represented by a line running from the large fiber system to central control mechanisms, which in turn project back to the gate control system. The T cells project to the entry cells of the action system. +, excitation; −, inhibition.
THE GATE CONTROL THEORY OF PAIN
The Gate Control beskriver hvordan stimulering av store nervefibre, f.eks. å blåse på sår, stryke på huden, osv., (mekanoreseptorer i huden) kan overdøve smertesignalene som kommer fra små nervefibre (nociceptive C-fibre). Gate Control teorien var den første som viste hvordan sentralnervesystemet kunne nedregulere smerte ovenifra og ned. Som inkluderer hjernens respons på signalene fra kroppen.
The final model, depicted in Figure 1(d), is the first theory of pain to incorporate the central control processes of the brain.
The gate control theory of pain11 proposed that the transmission of nerve impulses from afferent fibers to spinal cord transmission (T) cells is modulated by a gating mechanism in the spinal dorsal horn. This gating mechanism is influenced by the relative amount of activity in large- and small-diameter fibers, so that large fibers tend to inhibit transmission (close the gate) while small-fibers tend to facilitate transmission (open the gate).
When the output of the spinal T cells exceeds a critical level, it activates the Action System—those neural areas that underlie the complex, sequential patterns of behavior and experience characteristic of pain.
Psychological factors, which were previously dismissed as ‘reactions to pain’, were now seen to be an integral part of pain processing and new avenues for pain control by psychological therapies were opened.
BEYOND THE GATE
We believe the great challenge ahead of us is to understand brain function. Melzack and Casey13 made a start by proposing that specialized systems in the brain are involved in the sensory-discriminative, motivational-affective and cognitive-evaluative dimensions of subjective pain experience (Figure 2).
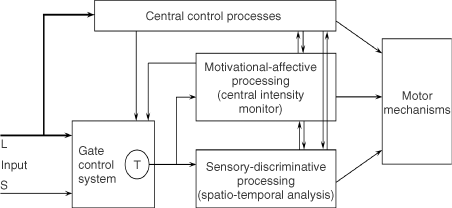
Figure 2. Conceptual model of the sensory, motivational, and central control determinants of pain. The output of the T (transmission) cells of the gate control system projects to the sensory-discriminative system and the motivational-affective system. The central control trigger is represented by a line running from the large fiber system to central control processes; these, in turn, project back to the gate control system, and to the sensory-discriminative and motivational-affective systems. All three systems interact with one another, and project to the motor system.
The newest version, the Short-Form McGill Pain Questionnaire-2,16 was designed to measure the qualities of both neuropathic and non-neuropathic pain in research and clinical settings.
In 1978, Melzack and Loeser17 described severe pains in the phantom body of paraplegic patients with verified total sections of the spinal cord, and proposed a central ‘pattern generating mechanism’ above the level of the section. This concept represented a revolutionary advance: it did not merely extend the gate; it said that pain could be generated by brain mechanisms in paraplegic patients in the absence of a spinal gate because the brain is completely disconnected from the cord. Psychophysical specificity, in such a concept, makes no sense; instead we must explore how patterns of nerve impulses generated in the brain can give rise to somesthetic experience.
Phantom Limbs and the Concept of a Neuromatrix
But there is a set of observations on pain in paraplegic patients that just does not fit the theory. This does not negate the gate theory, of course. Peripheral and spinal processes are obviously an important part of pain and we need to know more about the mechanisms of peripheral inflammation, spinal modulation, midbrain descending control, and so forth. But the data on painful phantoms below the level of total spinal cord section18,19 indicate that we need to go above the spinal cord and into the brain.
The cortex, Gybels and Tasker made amply clear, is not the pain center and neither is the thalamus.20 The areas of the brain involved in pain experience and behavior must include somatosensory projections as well as the limbic system.
First, because the phantom limb feels so real, it is reasonable to conclude that the body we normally feel is subserved by the same neural processes in the brain as the phantom; these brain processes are normally activated and modulated by inputs from the body but they can act in the absence of any inputs.
Second, all the qualities of experience we normally feel from the body, including pain, are also felt in the absence of inputs from the body; from this we may conclude that the origins of the patterns of experience lie in neural networks in the brain; stimuli may trigger the patterns but do not produce them.
Third, the body is perceived as a unity and is identified as the ‘self’, distinct from other people and the surrounding world. The experience of a unity of such diverse feelings, including the self as the point of orientation in the surrounding environment, is produced by central neural processes and cannot derive from the peripheral nervous system or spinal cord.
Fourth, the brain processes that underlie the body-self are ‘built-in’ by genetic specification, although this built-in substrate must, of course, be modified by experience, including social learning and cultural influences. These conclusions provide the basis of the conceptual model18,19,21 depicted in Figure 3.
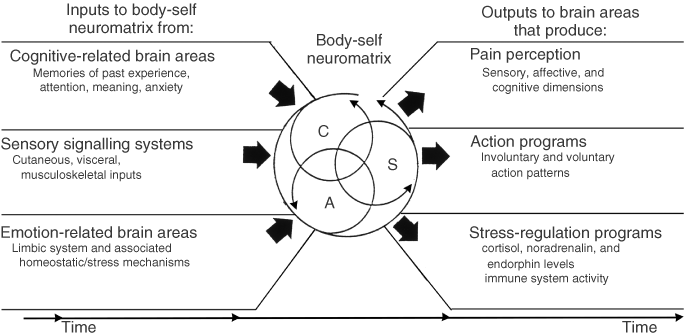
Figure 3. Factors that contribute to the patterns of activity generated by the body-self neuromatrix, which is comprised of sensory, affective, and cognitive neuromodules. The output patterns from the neuromatrix produce the multiple dimensions of pain experience, as well as concurrent homeostatic and behavioral responses.
Outline of the Theory
The anatomical substrate of the body-self is a large, widespread network of neurons that consists of loops between the thalamus and cortex as well as between the cortex and limbic system.18,19,21 The entire network, whose spatial distribution and synaptic links are initially determined genetically and are later sculpted by sensory inputs, is a neuromatrix. The loops diverge to permit parallel processing in different components of the neuromatrix and converge repeatedly to permit interactions between the output products of processing. The repeated cyclical processing and synthesis of nerve impulses through the neuromatrix imparts a characteristic pattern: the neurosignature. The neurosignature of the neuromatrix is imparted on all nerve impulse patterns that flow through it; the neurosignature is produced by the patterns of synaptic connections in the entire neuromatrix.
The neurosignature, which is a continuous output from the body-self neuromatrix, is projected to areas in the brain—the sentient neural hub—in which the stream of nerve impulses (the neurosignature modulated by ongoing inputs) is converted into a continually changing stream of awareness. Furthermore, the neurosignature patterns may also activate a second neuromatrix to produce movement, the action-neuromatrix .
The Body-Self Neuromatrix
The neuromatrix (not the stimulus, peripheral nerves or ‘brain center’) is the origin of the neurosignature; the neurosignature originates and takes form in the neuromatrix. Though the neurosignature may be activated or modulated by input, the input is only a ‘trigger’ and does not produce the neurosignature itself. The neuromatrix ‘casts’ its distinctive signature on all inputs (nerve impulse patterns) which flow through it.
The neuromatrix, distributed throughout many areas of the brain, comprises a widespread network of neurons which generates patterns, processes information that flows through it, and ultimately produces the pattern that is felt as a whole body.
Conceptual Reasons for a Neuromatrix
It is difficult to comprehend how individual bits of information from skin, joints, or muscles can all come together to produce the experience of a coherent, articulated body. At any instant in time, millions of nerve impulses arrive at the brain from all the body’s sensory systems, including the proprioceptive and vestibular systems. How can all this be integrated in a constantly changing unity of experience? Where does it all come together?
The neuromatrix, then, is a template of the whole, which provides the characteristic neural pattern for the whole body (the body’s neurosignature) as well as subsets of signature patterns (from neuromodules) that relate to events at (or in) different parts of the body
Alle har sett filmen The Matrix, sant? Spesielt scenen med «the spoonboy» er magisk: «Do not try to bend the spoon. That is impossible. Instead… only try to realize the truth» Neo: «What truth?». Spoonboy: «There is no spoon». Neo: «There is no spoon?». Spoonboy: «Then you´ll see, that it is not the spoon that bends, it is only your self». Dette har en direkte relasjon til smerteopplevelsen. Melzack forklarer:
Pain is not injury; the quality of pain experiences must not be confused with the physical event of breaking skin or bone. Warmth and cold are not ‘out there’; temperature changes occur ‘out there’, but the qualities of experience must be generated by structures in the brain. There are no external equivalents to stinging, smarting, tickling, itch; the qualities are produced by built-in neuromodules whose neurosignatures innately produce the qualities.
We do not learn to feel qualities of experience: our brains are built to produce them.
When all sensory systems are intact, inputs modulate the continuous neuromatrix output to produce the wide variety of experiences we feel. We may feel position, warmth, and several kinds of pain and pressure all at once. It is a single unitary feeling just as an orchestra produces a single unitary sound at any moment even though the sound comprises violins, cellos, horns, and so forth.
The experience of the body-self involves multiple dimensions—sensory, affective, evaluative, postural and many others.
To use a musical analogy once again, it is like the strings, tympani, woodwinds and brasses of a symphony orchestra which each comprise a part of the whole; each makes its unique contribution yet is an integral part of a single symphony which varies continually from beginning to end.
The output of the body neuromatrix is directed at two systems: (1) the neuromatrix that produces awareness of the output, and (2) a neuromatrix involved in overt action patterns. Just as there is a steady stream of awareness, there is also a steady output of behavior (including movements during sleep).
It is important to recognize that behavior occurs only after the input has been at least partially synthesized and recognized. For example, when we respond to the experience of pain or itch, it is evident that the experience has been synthesized by the body-self neuromatrix (or relevant neuromodules) sufficiently for the neuromatrix to have imparted the neurosignature patterns that underlie the quality of experience, affect and meaning. Most behavior occurs only after inputs have been analyzed and synthesized sufficiently to produce meaningful experience.
When we reach for an apple, the visual input has clearly been synthesized by a neuromatrix so that it has 3-dimensional shape, color and meaning as an edible, desirable object, all of which are produced by the brain and are not in the object ‘out there’. When we respond to pain (by withdrawal or even by telephoning for an ambulance), we respond to an experience that has sensory qualities, affect and meaning as a dangerous (or potentially dangerous) event to the body.
After inputs from the body undergo transformation in the body-neuromatrix, the appropriate action patterns are activated concurrently (or nearly so) with the neuromatrix for experience. Thus, in the action-neuromatrix, cyclical processing and synthesis produces activation of several possible patterns, and their successive elimination, until one particular pattern emerges as the most appropriate for the circumstances at the moment. In this way, input and output are synthesized simultaneously, in parallel, not in series. This permits a smooth, continuous stream of action patterns.
Another entrenched assumption is that perception of one’s body results from sensory inputs that leave a memory in the brain; the total of these signals becomes the body image. But the existence of phantoms in people born without a limb or who have lost a limb at an early age suggests that the neural networks for perceiving the body and its parts are built into the brain.18,19,27,28
Phantoms become comprehensible once we recognize that the brain generates the experience of the body. Sensory inputs merely modulate that experience; they do not directly cause it.
Pain and Neuroplasticity
Plasticity related to pain represents persistent functional changes, or ‘somatic memories,’29–31 produced in the nervous system by injuries or other pathological events.
Denervation Hypersensitivity and Neuronal Hyperactivity
Clinical neurosurgery studies reveal a similar relationship between denervation and CNS hyperactivity. Neurons in the somatosensory thalamus of patients with neuropathic pain display high spontaneous firing rates, abnormal bursting activity, and evoked responses to stimulation of body areas that normally do not activate these neurons.34,35
Furthermore, in patients with neuropathic pain, electrical stimulation of subthalamic, thalamic and capsular regions may evoke pain36 and in some instances even reproduce the patient’s pain.37–39
It is possible that receptive field expansions and spontaneous activity generated in the CNS following peripheral nerve injury are, in part, mediated by alterations in normal inhibitory processes in the dorsal horn. Within four days of a peripheral nerve section there is a reduction in the dorsal root potential, and therefore, in the presynaptic inhibition it represents.40 Nerve section also induces a reduction in the inhibitory effect of A-fiber stimulation on activity in dorsal horn neurons.41
The fact that amputees are more likely to develop phantom limb pain if there is pain in the limb prior to amputation30 raises the possibility that the development of longer term neuropathic pain also can be prevented by reducing the potential for central sensitization at the time of amputation.52,53
Pain and Psychopathology
Pain that is ‘nonanatomical’ in distribution, spread of pain to non-injured territory, pain that is said to be out of proportion to the degree of injury, and pain in the absence of injury have all, at one time or another, been used as evidence to support the idea that psychological disturbance underlies the pain. Yet each of these features of supposed psychopathology can now be explained by neurophysiological mechanisms that involve an interplay between peripheral and central neural activity.4,60
This raises the intriguing possibility that the intensity of pain at the site of an injury may be facilitated by contralateral neurite loss induced by the ipsilateral injury68—a situation that most clinicians would never have imagined possible.
Taken together, these novel mechanisms that explain some of the most puzzling pain symptoms must keep us mindful that emotional distress and psychological disturbance in our patients are not at the root of the pain. In fact, more often than not, prolonged pain is the cause of distress, anxiety, and depression.
Attributing pain to a psychological disturbance is damaging to the patient and provider alike; it poisons the patient-provider relationship by introducing an element of mutual distrust and implicit (and at times, explicit) blame. It is devastating to the patient who feels at fault, disbelieved and alone.
Pain and Stress
We are so accustomed to considering pain as a purely sensory phenomenon that we have ignored the obvious fact that injury does not merely produce pain; it also disrupts the brain’s homeostatic regulation systems, thereby producing ‘stress’ and initiating complex programs to reinstate homeostasis. By recognizing the role of the stress system in pain processes, we discover that the scope of the puzzle of pain is vastly expanded and new pieces of the puzzle provide valuable clues in our quest to understand chronic pain.69
However, it is important for the purpose of understanding pain to keep in mind that stress involves a biological system that is activated by physical injury, infection, or any threat to biological homeostasis, as well as by psychological threat and insult of the body-self.
When injury occurs, sensory information rapidly alerts the brain and begins the complex sequence of events to re-establish homeostasis. Cytokines are released within seconds after injury. These substances, such as gamma-interferon, interleukins 1 and 6, and tumor necrosis factor, enter the bloodstream within 1–4 min and travel to the brain. The cytokines, therefore, are able to activate fibers that send messages to the brain and, concurrently, to breach the blood–brain barrier at specific sites and have an immediate effect on hypothalamic cells. The cytokines together with evaluative information from the brain rapidly begin a sequence of activities aimed at the release and utilization of glucose for necessary actions, such as removal of debris, the repair of tissues, and (sometimes) fever to destroy bacteria and other foreign substances. Following severe injury, the noradrenergic system is activated: epinephrine is released into the blood stream and the powerful locus coeruleus/norepinephrine system in the brainstem projects information upward throughout the brain and downward through the descending efferent sympathetic nervous system. Thus, the whole sympathetic system is activated to produce readiness of the heart, blood vessels, and other viscera for complex programs to reinstate homeostasis.70,71
At the same time, the perception of injury activates the hypothalamic–pituitary–adrenal (HPA) system and the release of cortisol from the adrenal cortex, which inevitably plays a powerful role in determining chronic pain. Cortisol also acts on the immune system and the endogeneous opioid system. Although these opioids are released within minutes, their initial function may be simply to inhibit or modulate the release of cortisol. Experiments with animals suggest that their analgesic effects may not appear until as long as 30 min after injury.
Cortisol is an essential hormone for survival because it is responsible for producing and maintaining high levels of glucose for rapid response after injury or major threat. However, cortisol is potentially a highly destructive substance because, to ensure a high level of glucose, it breaks down the protein in muscle and inhibits the ongoing replacement of calcium in bone. Sustained cortisol release, therefore, can produce myopathy, weakness, fatigue, and decalcification of bone. It can also accelerate neural degeneration of the hippocampus during aging. Furthermore, it suppresses the immune system.
Estrogen increases the release of peripheral cytokines, such as gamma-interferon, which in turn produce increased cortisol. This may explain why more females than males suffer from most kinds of chronic pain as well as painful autoimmune diseases such as multiple sclerosis and lupus.72
Some forms of chronic pain may occur as a result of the cumulative destructive effect of cortisol on muscle, bone, and neural tissue. Furthermore, loss of fibers in the hippocampus due to aging reduces a natural brake on cortisol release which is normally exerted by the hippocampus. As a result, cortisol is released in larger amounts, producing a greater loss of hippocampal fibers and a cascading deleterious effect
The cortisol output by itself may not be sufficient to cause any of these problems, but rather provides the conditions so that other contributing factors may, all together, produce them.
The fact that several autoimmune diseases are also classified as chronic pain syndromes—such as Crohn’s disease, multiple sclerosis, rheumatoid arthritis, scleroderma, and lupus—suggests that the study of these syndromes in relation to stress effects and chronic pain could be fruitful. Immune suppression, which involves prolonging the presence of dead tissue, invading bacteria, and viruses, could produce a greater output of cytokines, with a consequent increase in cortisol and its destructive effects.
In some instances, pain itself may serve as a traumatic stressor.
Phantom Limb Pain
The cramping pain, however, may be due to messages from the action-neuromodule to move muscles in order to produce movement. In the absence of the limbs, the messages to move the muscles become more frequent and ‘stronger’ in the attempt to move the limb. The end result of the output message may be felt as cramping muscle pain. Shooting pains may have a similar origin, in which action-neuromodules attempt to move the body and send out abnormal patterns that are felt as shooting pain. The origins of these pains, then, lie in the brain.
Low-Back Pain
Protruding discs, arthritis of vertebral joints, tumors, and fractures are known to cause low back pain. However, about 60–70% of patients who suffer severe low back pain show no evidence of disc disease, arthritis, or any other symptoms that can be considered the cause of the pain. Even when there are clear-cut physical and neurological signs of disc herniation (in which the disc pushes out of its space and presses against nerve roots), surgery produces complete relief of back pain and related sciatic pain in only about 60% of cases.
A high proportion of cases of chronic back pain may be due to more subtle causes. The perpetual stresses and strains on the vertebral column (at discs and adjacent structures called facet joints) produce an increase in small blood vessels and fibrous tissue in the area.78 As a result, there is a release of substances that are known to produce inflammation and pain into local tissues and the blood stream; this whole stress cascade may be triggered repeatedly. The effect of stress-produced substances—such as cortisol and norepinephrine—at sites of minor lesions and inflammation could, if it occurs often and is prolonged, activate a neuromatrix program that anticipates increasingly severe damage and attempts to counteract it.
Fibromyalgia
An understanding of fibromyalgia has eluded us because we have failed to recognize the role of stress mechanisms in addition to the obvious sensory manifestations which have dominated research and hypotheses about the nature of fibromyalgia. Melzack’s interpretation of the available evidence is that the body-self neuromatrix’s response to stressful events fails to turn off when the stressor diminishes, so that the neuromatrix maintains a continuous state of alertness to threat. It is possible that this readiness for action produces fatigue in muscles, comparable to the fatigue felt by paraplegics in their phantom legs when they spontaneously make cycling movements.24 It is also possible that the prolonged tension maintained in particular sets of muscles produces the characteristic pattern of tender spots.
The persistent low-level stress (i.e., the failure of the stress response to cease) would produce anomalous alpha waves during deep sleep, greater feelings of fatigue, higher generalized sensitivity to all sensory inputs, and a low-level, sustained output of the stress-regulation system, reflected in a depletion of circulating cortisol.











Mye om huden relatert til smerte og nevropati! Mest relatert til biopsi, men mye kan knyttes til behandling også. Spesielt ved hemming av TRPV1.
http://www.hindawi.com/journals/nri/2013/329364/
This data has led to new insights into the potential pain mechanisms for various pain conditions including neuropathic pain (from small fiber neuropathies) and Complex Regional Pain Syndrome. The somatosensory neurons that innervate our skin constantly update our brains on the objects and environmental factors that surround us. Cutaneous sensory neurons expressing nociceptive receptors such as transient receptor potential vanilloid 1 channels and voltage-gated sodium channels are critical for pain transmission. Epidermal cells (such as keratinocytes, Langerhans cells, and Merkel cells) express sensor proteins and neuropeptides; these regulate the neuroimmunocutaneous system and participate in nociception and neurogenic inflammation.
The skin has homeostatic and immunologic barrier functions, but acts as a complex sensory organ as well [1]. The somatosensory neurons that innervate our skin constantly update our brains on the objects and environmental factors that surround us [2]. The neuroimmunocutaneous system (NICS) is responsible for the cutaneous sensations of touch, pressure, temperature, and pain. This sensory transduction occurs via primary afferent nerves following reciprocated signals between neuronal and nonneuronal skin cells of the NICS [1]. New data concerning peripheral pain mechanisms from within the skin have led to new insight into the potential pain mechanisms for various pain conditions including neuropathic pain syndromes such as diabetic neuropathy and Complex Regional Pain Syndrome.

In pain and neurogenic inflammation, TRPV1 is coexpressed on TRPA1-expressing sensory nerves; both integrate a variety of noxious stimuli [4]. Complex signaling pathways between cells of the NICS, such as keratinocytes, TRPV1-expressing nociceptors, and macrophages, lead to the release of neural growth factor (NGF), prostaglandins, opioids, proinflammatory cytokines, and chemokines [1]. These lead to sensitisation of the peripheral nerves by upregulating ionic channels and by inducing further spinal cord cytokine release [8].
2. Small Fiber Neuropathy (SFN)
Neuropathic pain arises as a direct consequence of a lesion or disease of the somatosensory system; it affects about 7% of the general population [10, 11].
Small fiber neuropathy is a neuropathy of the small nonmyelinated fibers and myelinated A delta fibers. Neuropathic pain occurs from small fiber neuropathy; small fiber neuropathy is caused by a wide variety of acquired and genetic disorders [12], many of which are treatable [13].
Diabetes mellitus is the most frequent underlying cause of SFN [14]. Other causes include toxic (e.g., alcohol), metabolic, immune-mediated, infectious, and hereditary causes.
About 60% of patients describe the painful sensation as spontaneous (burning, sunburn-like, paroxysmal, pruritic, and deep), with worsening at rest or during the night [12]; the sensation can be associated with thermal evoked pain (cold or warm) with or without allodynia, a painful response to a normally innocuous stimulus, and hyperalgesia, an increased response to a painful stimulus [12]. In addition there are negative sensory symptoms (thermal and pinprick hypoesthesia) that reflect peripheral deafferentation [19]. Sensation of cold feet is reported, though warm to touch. Thermal hypoesthesia with or without pinprick hypoesthesia has been detected in 40% of patients [20]; hyperalgesia and aftersensation have been detected in 10–20% of patients [12, 20].
Accumulating experimental and clinical evidence supports the hypothesis that Complex Regional Pain Syndrome type I (CRPS-I) might indeed be a small fiber neuropathy [25]. Most post-traumatic inflammatory changes observed in CRPS are mediated by two peptides, CGRP and substance P [26]. The activation of cutaneous nociceptors can induce retrograde depolarisation of small-diameter primary afferents, causing release of neuropeptides such as substance P and CGRP from sensory terminals in the skin.
A specific diagnostic test for small fiber neuropathy is a skin biopsy; this includes a count of the intraepidermal small nerve fibers (IENF) that cross the basal membrane. The loss of IENF can be reliably measured and is currently used to diagnose small fiber neuropathy (SFN) [17].

Skin biopsy is much less invasive and more practical than peripheral nerve biopsy. It is a safe and reliable tool for investigating nociceptive fibers in human epidermis and dermis [29]. It can be performed at any site of the body, with a disposable punch, using a sterile technique, and under local anesthesia (Figure 2) [29].
A recent study assessed the usefulness of skin biopsy in the assessment of 145 patients with suspected SFN [21]. In 59% of patients skin biopsy was abnormal in at least one site [21]. Patients with confirmed SFN were significantly more likely to have pain; they were more than twice as likely to respond to standard neuropathic pain medications [21]. A positive response to neuropathic pain medications was seen in 84% of patients with an abnormal skin biopsy compared to only 42% of those with a normal biopsy [21]. Skin biopsy has a relatively high yield in patients with sensory symptoms with no findings of mixed fiber neuropathy on clinical examination or on nerve conduction studies [21].
Along with neuronal and immunological systems, the skin plays a critical role in sensory transduction [1]. Further direct targeting of the skin with topical agents should be considered. The interaction of TRPV1 and TRPA1 channels in the skin in painful conditions needs further exploration. Second generation TRPV1 antagonists (without on-target side effects of hyperthermia and burn risk) are under development [6].
In Pain Medicine, the skin does indeed matter!
Om nerve compression syndrome, som sannsynligvis er årsaken til de fleste plager folk kommer til behandling for. Nevner hvordan nervevev påvirkes i løpet av timer, dager og uker. Nevner de 3 gradene av kompresjon og hvilke symptomer de gir.
http://ergo.berkeley.edu/docs/1999rempeljbjs.pdf
Nerve compression syndromes involve peripheral- nerve dysfunction that is due to localized interference of microvascular function and structural changes in the nerve or adjacent tissues.
When tissues are subjected to load or pressure, they deform and pressure gradients are formed, redistribut- ing the compressed tissue toward areas of lower pres- sure. Nerve compression syndromes usually occur at sites where the nerve passes through a tight tunnel formed by stiff tissue boundaries. The resultant con- fined space limits movement of tissue and can lead to sustained tissue pressure gradients. Space-occupying structures or lesions (for example, lumbrical muscles, tu- mors, and cysts) can cause nerve compression injury, as can conditions associated with accumulation of fluid (for example, pregnancy, congestive heart failure, and muscle compartment syndromes) or accumulation of extracellular matrix (for example, acromegaly, myx- edema hypothyroidism, and mucopolysaccharidosis)76.
Although nerve injuries related to vibration occur near the region of exposure, the symptoms may be manifest at another site, where the nerve may be constricted.
Other conditions, such as diabetes mellitus, may increase the likelihood that a compressed nerve will undergo a pathological response. In addition, there may be an in- flammatory reaction that may impair the normal gliding of the nerve.
Lying next to the myelinated nerve fibers are many nonmyelinated fibers associated with one Schwann cell. Myelinated and nonmyelinated nerve fibers are organized in bundles, called fascicles, which are surrounded by a strong membrane called the peri- neurial membrane, consisting of laminae of flattened cells.
Between the nerve fibers and their basal mem- brane is an intrafascicular connective tissue known as the endoneurium. The quantity of the connective-tissue components may vary between nerves and also along the length of the same nerve. For example, nerves lo- cated superficially in the limb or parts of the nerve that cross a joint contain a greater quantity of connective tissue, possibly as a response to repeated loading76.
The propagation of impulses in the nerve fibers as well as the communication and nutritional transport sys- tem in the neuron (axonal transport) requires an ade- quate energy supply. Therefore, the peripheral nerve contains a well developed microvascular system with vascular plexuses in all of its layers of connective tis- sue36,38. The vessels approach the nerve trunk segmen- tally and have a coiled configuration so that the vascular supply is not impaired during normal gliding or excur- sion of the nerve trunk. When the vessels reach the nerve trunk, they divide into branches that run longi- tudinally in various layers of the epineurium and they also form numerous collateral connections to vessels in the perineurial sheath. When the vessels pass through the perineurium into the endoneurium, which contains primarily capillaries, they often go through the perineu- rium obliquely, thereby constituting a possible valve mechanism36,38.
The perineurial layer and the endoneurial vessels play an important role in protecting the nerve fibers in the fascicles. The endoneurial milieu is protected by a blood-nerve barrier, and the tissue pressure in the fascicle (endoneurial fluid pressure) is slightly positive50.
The median and ulnar nerves may glide 7.3 and 9.8 millimeters, respectively, during full flexion and extension of the elbow, and the extent of excursion of these nerves just proximal to the wrist is even more pronounced (14.5 and 13.8 millimeters, respectively)90. In relation to the flexor retinaculum, the median nerve can move a maximum of 9.6 millimeters during wrist flexion and somewhat less during wrist extension; it also moves during motion of the fingers48.
Acute Effects of Nerve Compression (Effects within Hours)
In animal experiments, low-magnitude extraneural compression was noted to decrease intraneural micro- vascular flow, impair axonal transport, and alter nerve structure and function. Extraneural pressures of 2.7 kilo- pascals (twenty millimeters of mercury), induced with use of miniature inflatable cuffs, reduced epineurial ve- nule blood flow68. At pressures of 10.7 kilopascals (eighty millimeters of mercury), all intraneural blood flow ceased. Similarly, pressures of 4.0 kilopascals (thirty mil- limeters of mercury) inhibited both fast and slow ante- grade as well as retrograde axonal transport8.
In subjects with different blood pres- sures, the critical extraneural pressure threshold above which nerve function was blocked was 4.0 kilopascals (thirty millimeters of mercury) less than the diastolic pressure. This finding, combined with the observation that carpal tunnel syndrome may manifest with the treat- ment of hypertension17, provides additional support for an ischemic mechanism of acute nerve dysfunction.
Short-Term Effects of Nerve Compression (Effects within Days)
Com- pression of 4.0 kilopascals led to an elevated endoneu- rial pressure, which persisted for twenty-four hours after release of the cuff. Furthermore, the endoneurial pres- sures at twenty-four hours after release of the cuff increased with increasing durations of compression. His- tological examination demonstrated endoneurial edema in the nerves that had been subjected to eight hours of compression but not in those subjected to shorter dura- tions. Eight hours of compression led to an increase of the endoneurial pressures to levels that can reduce in- traneural blood flow51.
The study demonstrated that, af- ter low elevations of extraneural pressure for only two hours, endoneurial fluid pressures increased rapidly and the increases persisted for at least an additional twentyfour hours40. These effects probably are due to the in- creased vascular permeability of the epineurial and en- doneurial vessels after compression. Other studies have demonstrated that ischemia alters the structure of the endothelial and basement membranes over a similar time-frame2.
Long-Term Effects of Nerve Compression (Effects within Weeks)
Edema was visible in the sub- perineurial space within four hours in all compression subgroups, and it persisted for the entire duration of the study. Inflammation and fibrin deposits occurred within hours after compression, followed by prolifera- tion of endoneurial fibroblasts and capillary endothe- lial cells. Vigorous proliferation of fibrous tissue was noted within days, and marked fibrosis and sheets of fibrous tissue were seen extending to adjacent structures at twenty-eight days. Endoneurial invasion of mast cells and macrophages was noted, especially at twenty-eight days. Axonal degeneration was noted in the nerves sub- jected to 10.7 kilopascals of compression and, to a lesser extent, in those subjected to 4.0 kilopascals of compres- sion. It rarely was seen in the nerves subjected to 1.3 kilopascals of compression. Axonal degeneration was associated with the degree of endoneurial edema. De- myelination and Schwann-cell necrosis at seven and ten days was followed by remyelination at fourteen and twenty-eight days. Demyelination was prominent in the nerves subjected to 4.0 kilopascals of compres- sion and, to a lesser extent, in those subjected to1.3 kilo- pascals of compression.
The tension of the ligatures or the inner diameter of the tube generally was selected so that blood flow was not visibly restricted. The re- sponse of nerves to compression in these studies was similar to that in the experiments involving compression with a cuff. For example, the application of loose liga- tures around the sciatic nerve led to perineurial edema with proliferation of endothelial cells and demyelina- tion within the first few days, to proliferation of fibro- blasts and macrophages as well as degeneration of distal nerve fibers and the beginning of nerve sprouts within one week, to invasion by fibrous tissue and remyelina- tion at two weeks, to regeneration of nerve fibers as well as thickening of the perineurium and the vessel walls at six weeks, and to remyelination of distal nerve segments at twelve weeks73.
Applica- tion of silicone tubes with a wide internal diameter can induce increased expression of interleukin-1 and trans- forming growth factor beta-1 in the nerve cell bodies in the dorsal root ganglia, but the relevance of this finding remains to be clarified92. The limitations of these models are that (1) the effects of the tissue inflammatory reac- tion to the device (foreign-body reaction) usually are not considered but do occur29 and (2) it is not possible to measure or control the applied extraneural pressure. However, these observational studies provide some in- sight into the biological response of the nerve to chronic low-grade compression.
In a few case reports on patients in whom a nerve segment was resected, the nerve at the site of the injury was compared with a nerve at a site proximal or distal to the injury47,55,82. In each instance, there was thickening of the walls of the microvessels in the endo- neurium and perineurium as well as epineurial and peri- neurial edema, thickening, and fibrosis at the site of the injury. Thinning of the myelin also was noted, along with evidence of degeneration and regeneration of fibers. The patients in these reports had advanced stages of compression syndrome. Earlier in the course of the dis- ease, a segment of the nerve usually is compressed with disturbance of the microcirculation, which is restored immediately after transection of the flexor retinaculum. There is usually both an immediate and a delayed return of nerve function, indicating the importance of ischemia in the early stages of compression syndrome43.
The tissues that lie next to a nerve, within a confined space (for example, synovial tissue within the carpal tunnel), are more easily obtained and can provide infor- mation on the response of these tissues to compres- sion18,20,32,53,61,69,70,91.
The im- portant findings were increased edema and vascular sclerosis (endothelial thickening) in samples from the patients, who were between the ages of nineteen and seventy-nine years. Inflammatory cell infiltrates (lym- phocytes and histiocytes) were observed in only 10 per- cent (seventeen) of the 177 samples. Surprisingly, the prevalence of fibrosis (3 percent [five of 177]) was much lower than the prevalences of 33 percent (fifteen of forty-five) to 100 percent (twenty-one of twenty-one) reported in the other studies.
The initial symptoms of compres- sion of the median nerve at the wrist (carpal tunnel syndrome) usually are intermittent paresthesia and def- icits of sensation that occur primarily at night (stage I). These symptoms probably are due to changes in the intraneural microcirculation that are associated with some edema, which disappears during the day.
Progres- sive compression leads to more severe and constant symptoms that do not disappear during the day (stage II); these include paresthesia and numbness, impaired dexterity, and, possibly, muscle weakness. During this stage, the microcirculation may be altered throughout the day by edema and there may be morphological changes such as segmental demyelination.
In the final stage (stage III), there are more pronounced morpho- logical changes accompanied by degeneration of the nerve fibers; these changes manifest as constant pain with atrophy of the median-nerve-innervated thenar muscles and permanent sensory dysfunction.
In a study of the ulnar nerve at the elbow, localized areas of strain (nerve-stretching) of greater than 10 percent were observed in some cadav- eric arms83. A strain of 6 to 8 percent can limit blood flow in a nerve or can alter nerve function5,37,59.
Overview
First, elevated extraneural pressures can, within min- utes or hours, inhibit intraneural microvascular blood flow, axonal transport, and nerve function and also can cause endoneurial edema with increased intrafascicular pressure and displacement of myelin, in a dose-response manner. Pressures of 2.7 kilopascals (twenty millimeters of mercury) can limit epineurial blood flow, pressures of 4.0 kilopascals (thirty millimeters of mercury) can limit axonal transport and can cause nerve dysfunction and endoneurial edema, and pressures of 6.7 kilopascals (fifty millimeters of mercury) can alter the structure of myelin sheaths.
Second, on the basis of several animal models, it is apparent that low-magnitude, short-duration extraneu- ral pressure (for example, 4.0 kilopascals [thirty millime-
ters of mercury] applied for two hours) can initiate a process of nerve injury and repair and can cause struc- tural tissue changes that persist for at least one month.
The cascade of the bio- logical response to compression includes endoneurial edema, demyelination, inflammation, distal axonal de- generation, fibrosis, growth of new axons, remyelination, and thickening of the perineurium and endothelium. The degree of axonal degeneration is associated with the amount of endoneurial edema.
Third, in healthy people, non-neutral positions of the fingers, wrist, and forearm and loading of the fingertips can elevate extraneural pressure in the carpal tunnel in a dose-response manner. For example, fingertip pinch forces of five, ten, and fifteen newtons can elevate pres- sures to 4.0, 5.6, and 6.6 kilopascals
Fourth, in a rat model, exposure of the hindlimb to vibration for four to five hours per day for five days can cause intraneural edema, structural changes in my- elinated and unmyelinated fibers in the sciatic nerve, and functional changes both in nerve fibers and in non- neuronal cells.
Fifth, exposure to vibrating hand tools at work can lead to permanent nerve injury with structural neuronal changes in finger nerves as well as in the nerve trunks just proximal to the wrist. The relationships between the duration of exposure, the magnitude of the vibration, and structural changes in the nerve are unknown.
Om at vibrasjon på huden kan gjøre at C-fibre ikke reagerer så kraftig. Kan indikere en smertereduksjon.
http://link.springer.com/article/10.1007%2FBF01053477
Vibrostimulation of skin from the crus of the cat at frequencies of 10, 30, and 50/sec evokes a response of mechanoreceptors innervating group Aβ and Aδ fibers to the presentation of each vibrostimulus. The higher the frequency of vibrostimulation, the more clearly manifest are changes in on- and off-responses on a neurogram recorded from the n. saphenus branch. These changes are a result of a decrease in the number of impulses evoked by each vibratory beat. Mechanoreceptors innervated by fibers of group C are not excited as a result of vibrostimulation at these frequencies. After preliminary vibrostimulation, a portion of the mechanoreceptors innervated by C-fibers do not respond to presentation of a mechanical test stimulus (stretching of the skin); the higher the frequency of vibrostimulation, the smaller is the number of reacting receptors.
