Svært viktig studie med alt om smerte, fra Melzaks Body-Self Neuromatrix. Smerteforståelsens historie, fantomsmerter, hypersensitivitet, nervedegenerasjon, betennelser, Gate Control og Neuromatrix teori, m.m. Her forklares hvordan kroppsopplevelsen skapes i hjernen, selv uten noen input fra kroppen. Nevner også at smerte kan sette seg som et minne; somatic memory. Og vier mye plass til hvordan stress og kortisol bidrar til kroniske smerter, muskelsvikt og nedsatt immunsystem.
http://onlinelibrary.wiley.com/doi/10.1002/wcs.1201/full
Smerte har en funksjon i menneskekroppen som har utviklet seg i evolusjonen for å holde oss i live. Det gjør at vi tilpasser vår aktivitet så kroppen kan fokusere på helbredelse.
Pain has many valuable functions. It often signals injury or disease, generates a wide range of adaptive behaviors, and promotes healing through rest.
Men de siste 100-årenes (og foreløpige) forståelse av selve årsaken til smerte, hvordan den oppstår og hvordan den forsvinner, er basert på et mekanisk kroppsbilde som ikke tar hensyn til den subjektive smerteopplevelse. Melsaks arbeid viser oss hvordan vi snur dette og får en bedre og mer korrekt forståelse av smertefunksjonen:
Pain is a personal, subjective experience influenced by cultural learning, the meaning of the situation, attention, and other psychological variables. Pain processes do not begin with the stimulation of receptors. Rather, injury or disease produces neural signals that enter an active nervous system that (in the adult organism) is the substrate of past experience, culture, and a host of other environmental and personal factors.
Pain is not simply the end product of a linear sensory transmission system; it is a dynamic process that involves continuous interactions among complex ascending and descending systems. The neuromatrix theory guides us away from the Cartesian concept of pain as a sensation produced by injury, inflammation, or other tissue pathology and toward the concept of pain as a multidimensional experience produced by multiple influences.
Smerte er en helbredelsesfunksjon. Den hjelper oss å unngå truende situasjoner og sørger for at vi gir kroppen mulighet til å helbrede seg. Det er en naturlig og intelligent biologisk funksjon som i milliarder av år igjennom evolusjonen har sørget for at vi overlever så lenge som mulig.
We all know that pain has many valuable functions. It often signals injury or disease and generates a wide range of behaviors to end it and to treat its causes. Chest pain, for example, may be a symptom of heart disease, and may compel us to seek a physician’s help. Memories of past pain and suffering also serve as signals for us to avoid potentially dangerous situations. Yet another beneficial effect of pain, notably after serious injury or disease, is to make us rest, thereby promoting the body’s healing processes. All of these actions induced by pain—to escape, avoid, or rest—have obvious value for survival.
Smerteproblematikk har eksplodert de siste 20-30 årene og korsryggsmerter har overtatt plassen fra sult som den viktigste årsaken til ubehag blandt verdens befolkning. Melzak foreslår at vi bør se på kronisk smerte som en sykdom i seg selv, ikke som et symptom. En sykdom som følge av at nervesystemets alarm-mekanismer har slått seg vrang.
The pain, not the physical impairment, prevents them from leading a normal life. Likewise, most backaches, headaches, muscle pains, nerve pains, pelvic pains, and facial pains serve no discernible purpose, are resistant to treatment, and are a catastrophe for the people who are afflicted.
Pain may be the warning signal that saves the lives of some people, but it destroys the lives of countless others. Chronic pains, clearly, are not a warning to prevent physical injury or disease. They are the disease—the result of neural mechanisms gone awry.1–3
A BRIEF HISTORY OF PAIN
I smerteforskning og forståelse har vi, siden Descartes tid på 1600-tallet, beveget oss fra utsiden av kroppen igjennom det vi trodde var smerte-nervetråder, inn til ryggmargens «Gate Control», og nå, med The Neuromatrix, kommet opp til selve hjernen hvor vår opplevde virkelighet faktisk skapes. Først nå de siste årene har vi begynt å inkludere hjernens forskjellige funksjoner og dens eget «bilde» og opplevelse av kroppen. Tidligere ville pasienter som ikke ble bedre av kirurgi eller behandling bare bli avfeid av legene og heller sent til psykolog, hvor de heller ikke fikk noe spesifikk hjelp for smertene. Først nå, endelig, kan behandling av kronisk smerte inkludere større deler av mennesket som stemmer bedre overens med realiteten i både den subjektive opplevelsen og den vitenskapelige forklaringsmodellen.
The theory of pain we inherited in the 20th century was proposed by Descartes three centuries earlier. The impact of Descartes’ specificity theory was enormous. It influenced experiments on the anatomy and physiology of pain up to the first half of the 20th century (reviewed in Ref 4). This body of research is marked by a search for specific pain fibers and pathways and a pain center in the brain. The result was a concept of pain as a specific, direct-line sensory projection system. This rigid anatomy of pain in the 1950s led to attempts to treat severe chronic pain by a variety of neurosurgical lesions. Descartes’ specificity theory, then, determined the ‘facts’ as they were known up to the middle of the 20th century, and even determined therapy.
Specificity theory proposed that injury activates specific pain receptors and fibers which, in turn, project pain impulses through a spinal pain pathway to a pain center in the brain. The psychological experience of pain, therefore, was virtually equated with peripheral injury. In the 1950s, there was no room for psychological contributions to pain, such as attention, past experience, anxiety, depression, and the meaning of the situation.
Patients who suffered back pain without presenting signs of organic disease were often labeled as psychologically disturbed and sent to psychiatrists.
However, in none of these theories was there an explicit role for the brain other than as a passive receiver of messages. Nevertheless, the successive theoretical concepts moved the field in the right direction: into the spinal cord and away from the periphery as the exclusive answer to pain. At least the field of pain was making its way up toward the brain.
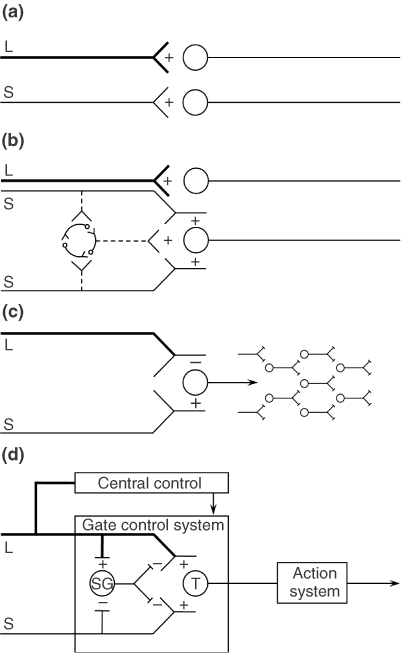
(D) Gate control theory. The large (L) and small (S) fibers project to the substantia gelatinosa (SG) and first central transmission (T) cells. The central control trigger is represented by a line running from the large fiber system to central control mechanisms, which in turn project back to the gate control system. The T cells project to the entry cells of the action system. +, excitation; −, inhibition.
THE GATE CONTROL THEORY OF PAIN
The Gate Control beskriver hvordan stimulering av store nervefibre, f.eks. å blåse på sår, stryke på huden, osv., (mekanoreseptorer i huden) kan overdøve smertesignalene som kommer fra små nervefibre (nociceptive C-fibre). Gate Control teorien var den første som viste hvordan sentralnervesystemet kunne nedregulere smerte ovenifra og ned. Som inkluderer hjernens respons på signalene fra kroppen.
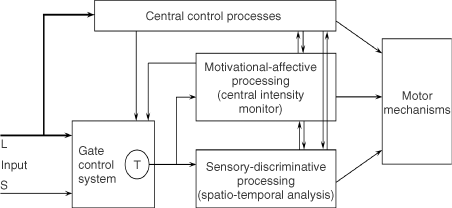
The final model, depicted in Figure 1(d), is the first theory of pain to incorporate the central control processes of the brain.
The gate control theory of pain11 proposed that the transmission of nerve impulses from afferent fibers to spinal cord transmission (T) cells is modulated by a gating mechanism in the spinal dorsal horn. This gating mechanism is influenced by the relative amount of activity in large- and small-diameter fibers, so that large fibers tend to inhibit transmission (close the gate) while small-fibers tend to facilitate transmission (open the gate).
When the output of the spinal T cells exceeds a critical level, it activates the Action System—those neural areas that underlie the complex, sequential patterns of behavior and experience characteristic of pain.
Psychological factors, which were previously dismissed as ‘reactions to pain’, were now seen to be an integral part of pain processing and new avenues for pain control by psychological therapies were opened.
BEYOND THE GATE
We believe the great challenge ahead of us is to understand brain function. Melzack and Casey13 made a start by proposing that specialized systems in the brain are involved in the sensory-discriminative, motivational-affective and cognitive-evaluative dimensions of subjective pain experience (Figure 2).
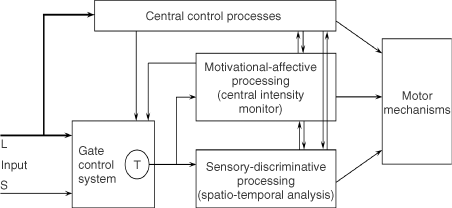
Figure 2. Conceptual model of the sensory, motivational, and central control determinants of pain. The output of the T (transmission) cells of the gate control system projects to the sensory-discriminative system and the motivational-affective system. The central control trigger is represented by a line running from the large fiber system to central control processes; these, in turn, project back to the gate control system, and to the sensory-discriminative and motivational-affective systems. All three systems interact with one another, and project to the motor system.
The newest version, the Short-Form McGill Pain Questionnaire-2,16 was designed to measure the qualities of both neuropathic and non-neuropathic pain in research and clinical settings.
In 1978, Melzack and Loeser17 described severe pains in the phantom body of paraplegic patients with verified total sections of the spinal cord, and proposed a central ‘pattern generating mechanism’ above the level of the section. This concept represented a revolutionary advance: it did not merely extend the gate; it said that pain could be generated by brain mechanisms in paraplegic patients in the absence of a spinal gate because the brain is completely disconnected from the cord. Psychophysical specificity, in such a concept, makes no sense; instead we must explore how patterns of nerve impulses generated in the brain can give rise to somesthetic experience.
Phantom Limbs and the Concept of a Neuromatrix
But there is a set of observations on pain in paraplegic patients that just does not fit the theory. This does not negate the gate theory, of course. Peripheral and spinal processes are obviously an important part of pain and we need to know more about the mechanisms of peripheral inflammation, spinal modulation, midbrain descending control, and so forth. But the data on painful phantoms below the level of total spinal cord section18,19 indicate that we need to go above the spinal cord and into the brain.
The cortex, Gybels and Tasker made amply clear, is not the pain center and neither is the thalamus.20 The areas of the brain involved in pain experience and behavior must include somatosensory projections as well as the limbic system.
First, because the phantom limb feels so real, it is reasonable to conclude that the body we normally feel is subserved by the same neural processes in the brain as the phantom; these brain processes are normally activated and modulated by inputs from the body but they can act in the absence of any inputs.
Second, all the qualities of experience we normally feel from the body, including pain, are also felt in the absence of inputs from the body; from this we may conclude that the origins of the patterns of experience lie in neural networks in the brain; stimuli may trigger the patterns but do not produce them.
Third, the body is perceived as a unity and is identified as the ‘self’, distinct from other people and the surrounding world. The experience of a unity of such diverse feelings, including the self as the point of orientation in the surrounding environment, is produced by central neural processes and cannot derive from the peripheral nervous system or spinal cord.
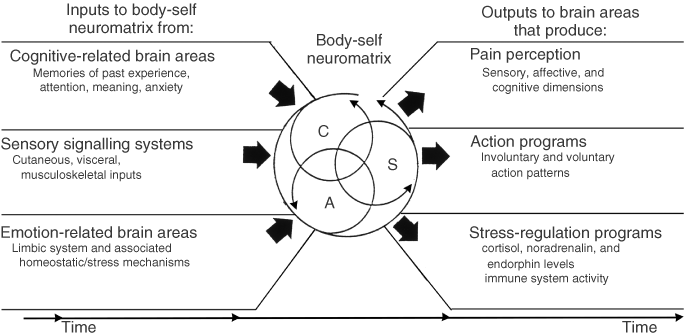
Fourth, the brain processes that underlie the body-self are ‘built-in’ by genetic specification, although this built-in substrate must, of course, be modified by experience, including social learning and cultural influences. These conclusions provide the basis of the conceptual model18,19,21 depicted in Figure 3.
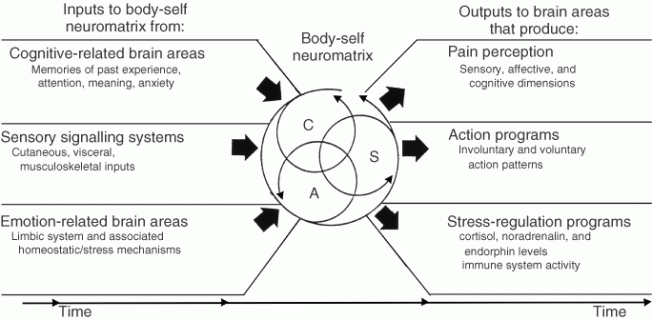
Figure 3. Factors that contribute to the patterns of activity generated by the body-self neuromatrix, which is comprised of sensory, affective, and cognitive neuromodules. The output patterns from the neuromatrix produce the multiple dimensions of pain experience, as well as concurrent homeostatic and behavioral responses.
Outline of the Theory
The anatomical substrate of the body-self is a large, widespread network of neurons that consists of loops between the thalamus and cortex as well as between the cortex and limbic system.18,19,21 The entire network, whose spatial distribution and synaptic links are initially determined genetically and are later sculpted by sensory inputs, is a neuromatrix. The loops diverge to permit parallel processing in different components of the neuromatrix and converge repeatedly to permit interactions between the output products of processing. The repeated cyclical processing and synthesis of nerve impulses through the neuromatrix imparts a characteristic pattern: the neurosignature. The neurosignature of the neuromatrix is imparted on all nerve impulse patterns that flow through it; the neurosignature is produced by the patterns of synaptic connections in the entire neuromatrix.
The neurosignature, which is a continuous output from the body-self neuromatrix, is projected to areas in the brain—the sentient neural hub—in which the stream of nerve impulses (the neurosignature modulated by ongoing inputs) is converted into a continually changing stream of awareness. Furthermore, the neurosignature patterns may also activate a second neuromatrix to produce movement, the action-neuromatrix .
The Body-Self Neuromatrix
The neuromatrix (not the stimulus, peripheral nerves or ‘brain center’) is the origin of the neurosignature; the neurosignature originates and takes form in the neuromatrix. Though the neurosignature may be activated or modulated by input, the input is only a ‘trigger’ and does not produce the neurosignature itself. The neuromatrix ‘casts’ its distinctive signature on all inputs (nerve impulse patterns) which flow through it.
The neuromatrix, distributed throughout many areas of the brain, comprises a widespread network of neurons which generates patterns, processes information that flows through it, and ultimately produces the pattern that is felt as a whole body.
Conceptual Reasons for a Neuromatrix
It is difficult to comprehend how individual bits of information from skin, joints, or muscles can all come together to produce the experience of a coherent, articulated body. At any instant in time, millions of nerve impulses arrive at the brain from all the body’s sensory systems, including the proprioceptive and vestibular systems. How can all this be integrated in a constantly changing unity of experience? Where does it all come together?
The neuromatrix, then, is a template of the whole, which provides the characteristic neural pattern for the whole body (the body’s neurosignature) as well as subsets of signature patterns (from neuromodules) that relate to events at (or in) different parts of the body
Alle har sett filmen The Matrix, sant? Spesielt scenen med «the spoonboy» er magisk: «Do not try to bend the spoon. That is impossible. Instead… only try to realize the truth» Neo: «What truth?». Spoonboy: «There is no spoon». Neo: «There is no spoon?». Spoonboy: «Then you´ll see, that it is not the spoon that bends, it is only your self». Dette har en direkte relasjon til smerteopplevelsen. Melzack forklarer:
Pain is not injury; the quality of pain experiences must not be confused with the physical event of breaking skin or bone. Warmth and cold are not ‘out there’; temperature changes occur ‘out there’, but the qualities of experience must be generated by structures in the brain. There are no external equivalents to stinging, smarting, tickling, itch; the qualities are produced by built-in neuromodules whose neurosignatures innately produce the qualities.
We do not learn to feel qualities of experience: our brains are built to produce them.
When all sensory systems are intact, inputs modulate the continuous neuromatrix output to produce the wide variety of experiences we feel. We may feel position, warmth, and several kinds of pain and pressure all at once. It is a single unitary feeling just as an orchestra produces a single unitary sound at any moment even though the sound comprises violins, cellos, horns, and so forth.
The experience of the body-self involves multiple dimensions—sensory, affective, evaluative, postural and many others.
To use a musical analogy once again, it is like the strings, tympani, woodwinds and brasses of a symphony orchestra which each comprise a part of the whole; each makes its unique contribution yet is an integral part of a single symphony which varies continually from beginning to end.
Action Patterns: The Action-Neuromatrix
The output of the body neuromatrix is directed at two systems: (1) the neuromatrix that produces awareness of the output, and (2) a neuromatrix involved in overt action patterns. Just as there is a steady stream of awareness, there is also a steady output of behavior (including movements during sleep).
It is important to recognize that behavior occurs only after the input has been at least partially synthesized and recognized. For example, when we respond to the experience of pain or itch, it is evident that the experience has been synthesized by the body-self neuromatrix (or relevant neuromodules) sufficiently for the neuromatrix to have imparted the neurosignature patterns that underlie the quality of experience, affect and meaning. Most behavior occurs only after inputs have been analyzed and synthesized sufficiently to produce meaningful experience.
When we reach for an apple, the visual input has clearly been synthesized by a neuromatrix so that it has 3-dimensional shape, color and meaning as an edible, desirable object, all of which are produced by the brain and are not in the object ‘out there’. When we respond to pain (by withdrawal or even by telephoning for an ambulance), we respond to an experience that has sensory qualities, affect and meaning as a dangerous (or potentially dangerous) event to the body.
After inputs from the body undergo transformation in the body-neuromatrix, the appropriate action patterns are activated concurrently (or nearly so) with the neuromatrix for experience. Thus, in the action-neuromatrix, cyclical processing and synthesis produces activation of several possible patterns, and their successive elimination, until one particular pattern emerges as the most appropriate for the circumstances at the moment. In this way, input and output are synthesized simultaneously, in parallel, not in series. This permits a smooth, continuous stream of action patterns.
Another entrenched assumption is that perception of one’s body results from sensory inputs that leave a memory in the brain; the total of these signals becomes the body image. But the existence of phantoms in people born without a limb or who have lost a limb at an early age suggests that the neural networks for perceiving the body and its parts are built into the brain.18,19,27,28
Phantoms become comprehensible once we recognize that the brain generates the experience of the body. Sensory inputs merely modulate that experience; they do not directly cause it.
Pain and Neuroplasticity
Plasticity related to pain represents persistent functional changes, or ‘somatic memories,’29–31 produced in the nervous system by injuries or other pathological events.
Denervation Hypersensitivity and Neuronal Hyperactivity
Clinical neurosurgery studies reveal a similar relationship between denervation and CNS hyperactivity. Neurons in the somatosensory thalamus of patients with neuropathic pain display high spontaneous firing rates, abnormal bursting activity, and evoked responses to stimulation of body areas that normally do not activate these neurons.34,35
Furthermore, in patients with neuropathic pain, electrical stimulation of subthalamic, thalamic and capsular regions may evoke pain36 and in some instances even reproduce the patient’s pain.37–39
It is possible that receptive field expansions and spontaneous activity generated in the CNS following peripheral nerve injury are, in part, mediated by alterations in normal inhibitory processes in the dorsal horn. Within four days of a peripheral nerve section there is a reduction in the dorsal root potential, and therefore, in the presynaptic inhibition it represents.40 Nerve section also induces a reduction in the inhibitory effect of A-fiber stimulation on activity in dorsal horn neurons.41
The fact that amputees are more likely to develop phantom limb pain if there is pain in the limb prior to amputation30 raises the possibility that the development of longer term neuropathic pain also can be prevented by reducing the potential for central sensitization at the time of amputation.52,53
Pain and Psychopathology
Pain that is ‘nonanatomical’ in distribution, spread of pain to non-injured territory, pain that is said to be out of proportion to the degree of injury, and pain in the absence of injury have all, at one time or another, been used as evidence to support the idea that psychological disturbance underlies the pain. Yet each of these features of supposed psychopathology can now be explained by neurophysiological mechanisms that involve an interplay between peripheral and central neural activity.4,60
This raises the intriguing possibility that the intensity of pain at the site of an injury may be facilitated by contralateral neurite loss induced by the ipsilateral injury68—a situation that most clinicians would never have imagined possible.
Taken together, these novel mechanisms that explain some of the most puzzling pain symptoms must keep us mindful that emotional distress and psychological disturbance in our patients are not at the root of the pain. In fact, more often than not, prolonged pain is the cause of distress, anxiety, and depression.
Attributing pain to a psychological disturbance is damaging to the patient and provider alike; it poisons the patient-provider relationship by introducing an element of mutual distrust and implicit (and at times, explicit) blame. It is devastating to the patient who feels at fault, disbelieved and alone.
Pain and Stress
We are so accustomed to considering pain as a purely sensory phenomenon that we have ignored the obvious fact that injury does not merely produce pain; it also disrupts the brain’s homeostatic regulation systems, thereby producing ‘stress’ and initiating complex programs to reinstate homeostasis. By recognizing the role of the stress system in pain processes, we discover that the scope of the puzzle of pain is vastly expanded and new pieces of the puzzle provide valuable clues in our quest to understand chronic pain.69
However, it is important for the purpose of understanding pain to keep in mind that stress involves a biological system that is activated by physical injury, infection, or any threat to biological homeostasis, as well as by psychological threat and insult of the body-self.
When injury occurs, sensory information rapidly alerts the brain and begins the complex sequence of events to re-establish homeostasis. Cytokines are released within seconds after injury. These substances, such as gamma-interferon, interleukins 1 and 6, and tumor necrosis factor, enter the bloodstream within 1–4 min and travel to the brain. The cytokines, therefore, are able to activate fibers that send messages to the brain and, concurrently, to breach the blood–brain barrier at specific sites and have an immediate effect on hypothalamic cells. The cytokines together with evaluative information from the brain rapidly begin a sequence of activities aimed at the release and utilization of glucose for necessary actions, such as removal of debris, the repair of tissues, and (sometimes) fever to destroy bacteria and other foreign substances. Following severe injury, the noradrenergic system is activated: epinephrine is released into the blood stream and the powerful locus coeruleus/norepinephrine system in the brainstem projects information upward throughout the brain and downward through the descending efferent sympathetic nervous system. Thus, the whole sympathetic system is activated to produce readiness of the heart, blood vessels, and other viscera for complex programs to reinstate homeostasis.70,71
At the same time, the perception of injury activates the hypothalamic–pituitary–adrenal (HPA) system and the release of cortisol from the adrenal cortex, which inevitably plays a powerful role in determining chronic pain. Cortisol also acts on the immune system and the endogeneous opioid system. Although these opioids are released within minutes, their initial function may be simply to inhibit or modulate the release of cortisol. Experiments with animals suggest that their analgesic effects may not appear until as long as 30 min after injury.
Cortisol is an essential hormone for survival because it is responsible for producing and maintaining high levels of glucose for rapid response after injury or major threat. However, cortisol is potentially a highly destructive substance because, to ensure a high level of glucose, it breaks down the protein in muscle and inhibits the ongoing replacement of calcium in bone. Sustained cortisol release, therefore, can produce myopathy, weakness, fatigue, and decalcification of bone. It can also accelerate neural degeneration of the hippocampus during aging. Furthermore, it suppresses the immune system.
Estrogen increases the release of peripheral cytokines, such as gamma-interferon, which in turn produce increased cortisol. This may explain why more females than males suffer from most kinds of chronic pain as well as painful autoimmune diseases such as multiple sclerosis and lupus.72
Some forms of chronic pain may occur as a result of the cumulative destructive effect of cortisol on muscle, bone, and neural tissue. Furthermore, loss of fibers in the hippocampus due to aging reduces a natural brake on cortisol release which is normally exerted by the hippocampus. As a result, cortisol is released in larger amounts, producing a greater loss of hippocampal fibers and a cascading deleterious effect
The cortisol output by itself may not be sufficient to cause any of these problems, but rather provides the conditions so that other contributing factors may, all together, produce them.
The fact that several autoimmune diseases are also classified as chronic pain syndromes—such as Crohn’s disease, multiple sclerosis, rheumatoid arthritis, scleroderma, and lupus—suggests that the study of these syndromes in relation to stress effects and chronic pain could be fruitful. Immune suppression, which involves prolonging the presence of dead tissue, invading bacteria, and viruses, could produce a greater output of cytokines, with a consequent increase in cortisol and its destructive effects.
In some instances, pain itself may serve as a traumatic stressor.
Phantom Limb Pain
The cramping pain, however, may be due to messages from the action-neuromodule to move muscles in order to produce movement. In the absence of the limbs, the messages to move the muscles become more frequent and ‘stronger’ in the attempt to move the limb. The end result of the output message may be felt as cramping muscle pain. Shooting pains may have a similar origin, in which action-neuromodules attempt to move the body and send out abnormal patterns that are felt as shooting pain. The origins of these pains, then, lie in the brain.
Low-Back Pain
Protruding discs, arthritis of vertebral joints, tumors, and fractures are known to cause low back pain. However, about 60–70% of patients who suffer severe low back pain show no evidence of disc disease, arthritis, or any other symptoms that can be considered the cause of the pain. Even when there are clear-cut physical and neurological signs of disc herniation (in which the disc pushes out of its space and presses against nerve roots), surgery produces complete relief of back pain and related sciatic pain in only about 60% of cases.
A high proportion of cases of chronic back pain may be due to more subtle causes. The perpetual stresses and strains on the vertebral column (at discs and adjacent structures called facet joints) produce an increase in small blood vessels and fibrous tissue in the area.78 As a result, there is a release of substances that are known to produce inflammation and pain into local tissues and the blood stream; this whole stress cascade may be triggered repeatedly. The effect of stress-produced substances—such as cortisol and norepinephrine—at sites of minor lesions and inflammation could, if it occurs often and is prolonged, activate a neuromatrix program that anticipates increasingly severe damage and attempts to counteract it.
Fibromyalgia
An understanding of fibromyalgia has eluded us because we have failed to recognize the role of stress mechanisms in addition to the obvious sensory manifestations which have dominated research and hypotheses about the nature of fibromyalgia. Melzack’s interpretation of the available evidence is that the body-self neuromatrix’s response to stressful events fails to turn off when the stressor diminishes, so that the neuromatrix maintains a continuous state of alertness to threat. It is possible that this readiness for action produces fatigue in muscles, comparable to the fatigue felt by paraplegics in their phantom legs when they spontaneously make cycling movements.24 It is also possible that the prolonged tension maintained in particular sets of muscles produces the characteristic pattern of tender spots.
The persistent low-level stress (i.e., the failure of the stress response to cease) would produce anomalous alpha waves during deep sleep, greater feelings of fatigue, higher generalized sensitivity to all sensory inputs, and a low-level, sustained output of the stress-regulation system, reflected in a depletion of circulating cortisol.




 h, generally starting at 10:00
h, generally starting at 10:00